Descargado 76 veces



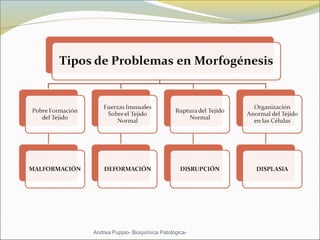
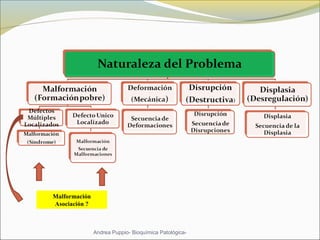


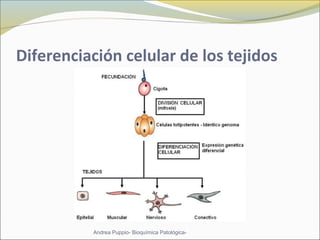

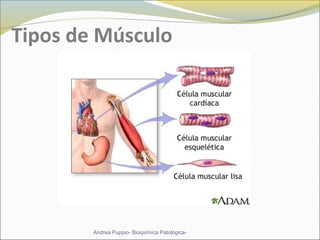



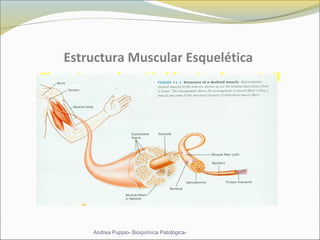

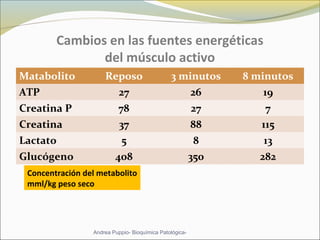












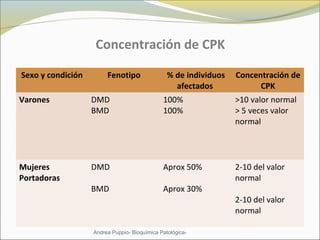







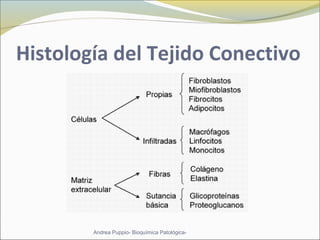




















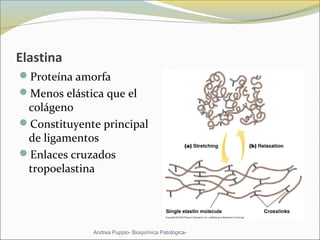

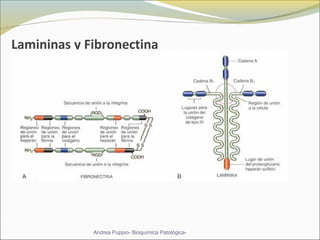

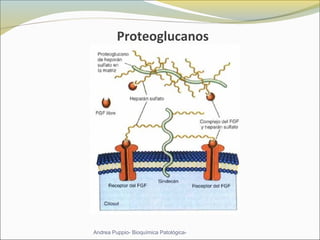

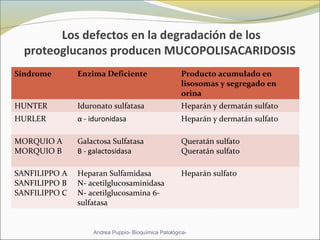







































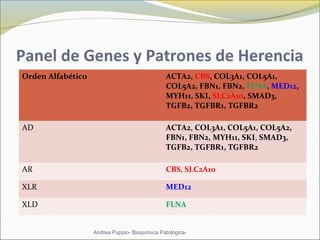





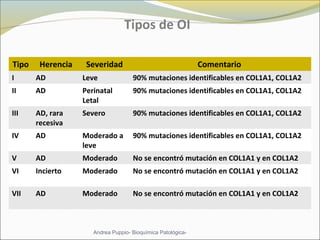
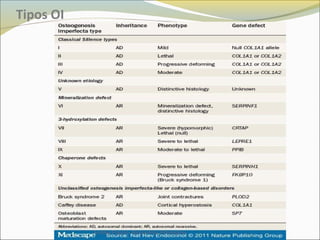
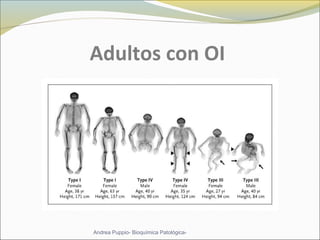
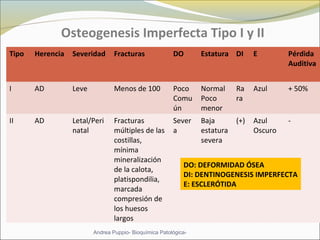
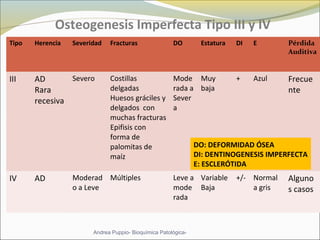
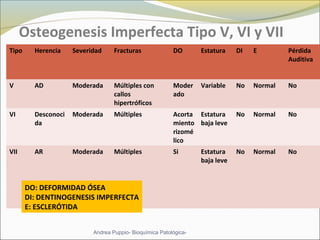



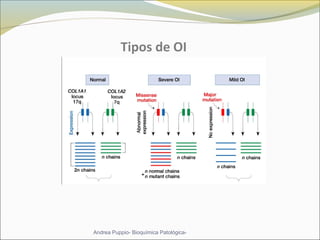
























Este documento describe las características del tejido muscular esquelético y las distrofias musculares de Duchenne y Becker. Explica que el tejido muscular esquelético está compuesto de miofibrillas y sarcómeros, y que las distrofias de Duchenne y Becker son enfermedades hereditarias causadas por mutaciones en el gen de la distrofina que codifica para la proteína distrofina y afectan principalmente al músculo y corazón. También describe los síntomas, diagnóstico y tratamiento de ambas enfermedades



































































