
Recomendados
Más contenido relacionado
Similar a ENDOCRINOLOGIA PARA EL ADULTO Y COMPLICACIONES
Similar a ENDOCRINOLOGIA PARA EL ADULTO Y COMPLICACIONES (20)
Más de MelacitoDess
Más de MelacitoDess (12)
Último
Último (20)
ENDOCRINOLOGIA PARA EL ADULTO Y COMPLICACIONES
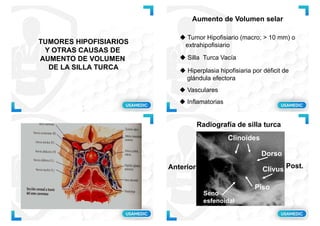
- 1. TUMORES HIPOFISIARIOS Y OTRAS CAUSAS DE AUMENTO DE VOLUMEN DE LA SILLA TURCA Aumento de Volumen selar Tumor Hipofisiario (macro; > 10 mm) o extrahipofisiario Silla Turca Vacía Vasculares Hiperplasia hipofisiaria por déficit de glándula efectora Inflamatorias Radiografía de silla turca Dorso Clivus Clinoides Piso Seno esfenoidal Anterior Post.
- 2. MACROADENOMA HIPOFISIARIO >1 cm EFECTO DE MASA Manifestaciones de los tumores selares Cefalea Alteración de quiasma (superior) Seno esfenoidal (inferior) Pares craneanos III, IV, VI (lateral) Neurohipófisis (posterior) HIPERSECRECION HORMONAL Prolactina Hormona Crecimiento ACTH Gonadotropinas DAÑO HIPOFISIARIO Hipopituitarismo Patogenia de los tumores hipofisiarios Generalmente son adenomas de crecimiento lento, carentes de elementos de malignidad, mayoritariamente esporádicos Patogenia Alteración genes: ganancia de función supresores de crecimiento tumoral responsables de apoptosis celular Distribución aproximada de Tumores selares Gonadotrópicos y No Func. 34% PRL 40% GH 15% ACTH 10% TSH 1%
- 3. ESCESO DE HORMONA DE CRECIMIENTO EXCESO DEFECTO gigantismo enanismo Pauline Musters 59 cm (Ossendrecht, Holanda, 1876-1895) Robert Wadlow 2,72 m (Ilinois, USA 1918-1940) Las alteraciones en la hormona de crecimiento durante la infancia producen gigantismo o enanismo Hormona de Crecimiento Transporte y Metabolismo 50% unida a proteínas plasmáticas se une a un fragmento de porción extracelular del receptor de GH: GH-BP vida media: 20-30 minutos producción diaria: 0,2-1 mg/día en el adulto secreción pulsátil con predominio nocturno
- 4. Factores en Secreción de GH Factores Estimulación Inhibición Nutrientes hipoglicemia Hiperglicema desnutrición proteica dism. de AGL Obesidad aum. de AGL Arginina Otros ejercicio sueño stress Medición de GH Edad: 5 -20 años: 5- 8 ng/ml 20-40 años: 0,2-3 ng/ml 40- 70 años: 0,2- 2 ng/ml Tests de estímulo: arginina- hipoglicemia- insulina- glucagón Tests de Inhibición: administración de glucosa oral HIPERPRODUCCIÓN DE GH ALTERACIONES METABOLICAS Metabolismo Fisiopatología Alt.Biolog. Relación P/Ca absorción. intestinal calcio Normo calcemia reab.tubular P hiperfosfemia reab.tubular Ca hipercalciuria Glucídico Antagonismo con Insulina Resistencia Insulínica gluconeogénesis Int. H de C Hiperproducción de GH. Alteraciones Metabólicas Metabolismo Fisiopatología Alt. Biologica lipídico Lipólisis: mobilización AGL de tej.adiposo AGL circulantes proteico Anabolismo: ingreso de aac. a tejidos Balance nitrogenado + Hidro- electrolítico reab. Tubular de Na LEC con hipervolemia
- 5. Prevalencia (millón hab): 38-60 casos Incidencia (millón hab): 2,8 4 nuevos casos/año Edad: 40-50 años, igual para ambos sexos Aumento de la morbimortalidad: Enfermedad Vascular: 50% (HTA, Diabetes, Hiperinsulinemia, Coronariopatía) Enfermedad Respiratoria: 22% Cáncer?: 16% Acromegalia Acromegalia. Cuadro Clínico Síntomas o signos % ensanchamiento óseo manos-pies 100 crecimiento partes blandas 100 hiperhidrosis 88 fatiga 87 de peso 73 parestesias 70 artralgias 69 fotofobia 46 papilomas cutáneos 45 Pólipos premalignos NM colon 3 veces mas riesgo Acromegalia Cuadro Clínico Manifestaciones < frecuentes (10-30 %) Hirsutismo Bocio Acanthosis nigricans Hipertensión arterial Cardiomegalia Litiasis renal
- 6. Acromegalia Cuadro Clínico Alteración de otras funciones endocrinas: % hiperinsulinemia con IR 70 intolerancia a carbohidratos 50 oligoamenorrea/amenorrea 60 líbido-impotencia 46 hipotiroidismo 13 galactorrea 13 Acromegalia. Cuadro Clínico Manifestaciones Locales. Síndrome Tumoral: % tamaño selar 90 cefalea 65 trastornos campo visual 20 0 20 40 60 80 100 Defecto campo visual Hipertensión Hipertricosis Falla cardíaca congestiva, arritmia Problemas dentales, nasales Bocio Intolerancia a la glucosa, diabetes Artropatía Disfunción erectil, disminución de libido Oligo-amenorrea, infertilidad Cefalea Debilidad, letargia Acroparestesia, S. tunel carpiano Sudoración Agrandamiento acral, tej. Blandos y rasgos faciales % Acromegalia: impacto sobre la sobrevida General population All acromegaly Acromegaly + diabetes Acromegaly + cardiac disease Life expectancy 10 years Length of survival (years) Adapted from Rajascorya C,et all. Clin Endocrinol (Oxl). 1994;41;95.
- 7. Acromegalia. Causas 1)T. hipofisiario productor de GH > 98 % correlación entre secreción de GH y tamaño TU pacientes jóvenes tienden a tener TU de > tamaño al momento del diag. 75 % son MACROADENOMAS tardan en promedio 7- 10 años en hacer el diagnóstico 2)Secreción de GH-RH hipotalámico < 1 % 3)Producción ectópica de GH < 1% DIAGNÓSTICO DE ACROMEGALIA 1) CLINICO 2) LABORATORIO: -certificar diagnóstico - estudiar otros ejes hipofisiarios -estudio de otros TU endocrinos (MEN) 3) IMÁGENES: -tamaño tumoral -sindrome tumoral IMAGENES Rx simple Silla turca TAC RMI Microadenoma Macroadenoma (Invasión a senos cavernosos, esfenoidal) NEUROOFTALMOLOGICO Campo visual con cúpula de Goldman Campo visual computarizado Acromegalia Diagnóstico por Imágenes Acromegalia Microadenomas
- 8. Acromegalia Macroadenomas Diagnóstico de Acromegalia. Laboratorio 1) inespecíficos: hiperfosfemia- hipercalciuria- hiperglicemia ayunas o postcarga de glucosa- enzimas hepáticas alteradas 2)específico: a) prueba de supresión con estímulo fisiológico: (75g) respuesta normal < 2 ug/l(RIA) y aumento de IGF-1 para valor normal según edad-sexo acromegalia: > 5 ng/ml Acromegalia -diagnostico Puede existir respuestas paradójicas de GH a estímulos hipotalámicos que en condiciones normales no la estimulan: TRH (80%) y GNRH (10-155). En acromegalicos la bromocriptina suele inhibir secreción de GH ACROMEGALIA. TRATAMIENTO OBJETIVOS: 1) niveles de GH a rangos normales(<2ug/l) 2) estabilización o del tamaño tumoral 3) preservación de otros ejes hipofisiarios QUIRURGICO RADIOTERAPIA MEDICO
- 9. Acromegalia. Tratamiento QUIRURGICO: cirugía transesfenoidal curación- mejoría ( partes blandas) respuesta según valores preop. < 40 ng//ml: 75 % de curación > 40 ng/ml : 35 % de curación complicaciones postop.: hipopituitarismo- rinorrea LCR-hemorragia RADIOTERAPIA: respuesta lenta complicaciones: NECROSIS TU con apoplejía hipopituitarismo: 50 % en 10 años Resección de tumor hipofisiario por vía transesfenoidal Acromegalia- Cirugía La tasa de curación es mayor cuando es un microadenoma (90%) comparado con un macroadenoma (20-40%) Se produce hipopituitarismo en un 10- 20% de los casos con macroadenomas Tratamiento médico de la acromegalia Estímulo de receptores de somatostatina en el tumor hipofisiario Análogos de somatostatina: Octreotide, Lanreotide Normalizan GH e IGF1 en el 50% con regresión de tumor en 30-50% Bloqueo del receptor de GH en los órganos efectores Pegvisomant: normaliza IGF1 en 90%
- 10. Tratamiento medico Los agonistas dopaminergicos en acromegalicos suelen inhibir secreción GH más no en sujetos sanos pero las dosis que se usan son mayores que las que se usan en prolactinomas. Es un tratamiento de segunda elección Radioterapia Indicación principal: post cirugía incompleta Como terapia primaria es más eficaz si los valores basales de GH son < 30 ng/mL Limitaciones: Efecto tardío y lento Alta incidencia de hipopituitarismo Cefalea, fatigabilidad, alts. Memoria Radionecrosis ENANISMO HIPOFISIARIO déficit GH 25-35%: Lesión hipofisiaria (displasia septooptica, holoprosencefalia, histiocitosis, craneofariongioma) la mayoría idiopáticos Si el déficit es congénito se manifiesta a las 6-12 meses de edad, talla y peso al nacer normal. Si es adquirido se detiene el crecimiento después de un crecimiento normal. Fenotipo: obesidad troncular, frente amplia abombada, mejillas redondeadas voz aguda y chillona DEFICIT DE GH NIÑO Déficit de GH Los niveles de GH son indetectables . Se realiza prueba funcional hipoglicemia insulinica o test de clonidina, con valores de GH superiores a 10/ug/l o 2 pruebas con valores menores a 5 ug/l Tratamiento GH sintética con una aceleración de crecimiento normal . Si hay insensibilidad a GH se usa IGF-1 recombinante (síndrome de Laron)
- 11. DEFICIT DE GH ADULTO Los tumores hipofisiarios tienen hipopituitarismo en el 80% de los casos al momento del dx y el 50% tienen déficit de GH, gonadotrofinas o cortisol Síntomas: aumento de la grasa corporal, disminución de la masa muscular, disminución de fuerza física , disminución de sudoración disminución de la vitalidad alteraciones psicológicas y de la calidad de vida Diagnóstico Hipoglicemia insulinica: la incapacidad de llegar a un pico superior a 3 ng/ml o un IGF-1 basal (solo en un 60%) de pacientes El tratamiento mejora la calidad de vida, corrige las alteraciones de composición corporal recupera masa ósea y muscular TUMORES PRODUCTORES DE PROLACTINA: PROLACTINOMAS LACTOTROPO DOPAMINA (-) HIPOTÁLAMO
- 12. Regulación de la secreción de Prolactina Secreción es inhibida tónicamente por la dopamina hipotalámica Estimulan la PRL: TRH hipotalámica, succión o manipulación del pezón, y los estrógenos (embarazo) VN: <20 ng/ml Causas de Hiperprolactinemia Prolactinomas (y acromegalia) Drogas: bloqueadores del receptor de dopamina (fenotiacinas, sulpiride) Hipotiroidismo Embarazo Interrupción del tallo hipot.-hipófisis I. Renal Crónica, SOP Idiopáticas Causas de Hiperprolactinemia Enfermedades Hipotalámicas Tumores, Metástasis, Craneofaringeoma, Glioma, Hamartoma, Quistes Enf. Infiltrativas (Sarcoidosis, Tuberculosis, Histiocitosis X) Radiación cerebral Causas de Hiperprolactinemia Enfermedades hipofisarias Prolactinoma Acromegalia Enfermedad de Cushing Gonadotrofinoma Tumores no secretorios Sección del tallo hipofisario Silla turca vacía Otros tumores (MTS, Meningioma, Germinoma) Enf. Infiltrativas (Sarcoidosis, TBC)
- 13. Causas de Hiperprolactinemia Drogas Antagonistas dopaminérgicos (sulpirida, clorpromazida, flufenazina, haloperidol, domperidona, metoclopramida) Otras (reserpina, metildopa, verapamil, estrógenos, opiáceos, cimetidina) General Hipotiroidismo Insuficiencia renal crónica Cirrosis Lesiones de pared torácica Causas de Hiperprolactinemia Fisiológicas Estrés Embarazo Lactancia Período neonatal Ejercicio Sueño Relación sexual Idiopática HIPERPROLACTINEMIA EN LA MUJER Síndrome de Amenorrea-Galactorrea Chiari-Frommel (postparto), Ahumada-Del Castillo ó Forbes-Albright Oligo/amenorrea Galactorrea Infertilidad Cefalea Alteración del campo visual Oftalmoplejía Retraso puberal Hipogonadismo hiupogonadotrófico HIPERPROLACTINEMIA EN EL VARON SINTOMAS Y SIGNOS Disminución de libido y erección Cefalea Ginecomastia - Galactorrea Alteración del campo visual Oligospermia - Astenospermia Oftalmoplejía Disminución volumen del ejaculado Retraso puberal Hipogonadismo hipogonadotrófico
- 14. PROLACTINOMA Es el adenoma hipofisario más frecuente El cuadro clínico depende de: sexo edad de presentación tiempo de hiperprolactinemia tamaño del tumor Prolactinomas: Generalidades Etiología: Tumores monoclonales; origen de novo Frecuencia: Desconocida. En autopsias ± 25% presentan microadenomas (40% con tinción PRL +) Evolución: - Microadenomas: la mayoría tiende a mantener su tamaño en el tiempo. Menos del 5% crece durante el embarazo. - Macroadenomas: tienen riesgo de crecer, especialmente en el embarazo (25%). PROLACTINOMA Microadenoma (< 10 mm) Macroadenoma (> 10 mm) Intraselar Expansión supraselar, seno cavernoso, seno esfenoidal Invasivo Prolactinomas: Clínica 1. Hipogonadismo hipogonadotropo - oligomenorrea ó amenorrea secundaria - rara vez pubertad retrasada - deterioro libido, sequedad vaginal; - Impotencia en varones 2.Galactorrea (30-80%) (casi exclusivo en mujeres 3.Cefalea o alteraciones visuales (en tumores > 10mm) 4. Osteopenia por hipogonadismo
- 15. Prolactinomas: Tratamiento Primera opción: Tratamiento médico -Agonistas Dopaminérgicos (Cabergolina, Bromocriptina) Tratamiento Quirúrgico en: Fracaso tratamiento médico -Macroadenomas en mujeres que buscan fertilidad -Macroadenomas complicados -Intolerancia a drogas Prolactinomas Microprolactinomas: no siempre se trata, pero los macro si En los micro: mujeres con deseo de embarazo, hipogonadismo severo con alto riesgo de osteoporoisis galactorrea molesta disminución de la libido. Nombre Genérico Dosis Habitual Bromocriptina 2.5 mg-15 mg (2/día) Cabergolina 0.5-3 mg, 1-2 veces/semana Pergolide 0.05-0.2 mg/día Quinagolide 0.075-0.4 mg/día TRATAMIENTO MÉDICO DEL PROLACTINOMA
- 16. OTRAS LESIONES HIPOFISIARIAS Incidentalomas hipofisiarios Frecuencia: 61/1000 (en autopsias) (6%) Quistes 37 Adenomas 18 Hiperplasia 2 Infarto 2 Hemorragia 2 Teramoto et al., Radiology 1994; 193: 161 La mayoría de las hormonas hipofisiarias dependen de factores liberadores hipotalámicos. La excepción es la PRL que es inhibida tónicamente por la dopamina hipotalámica. La Sección de Tallo produce déficit de las hormonas de la adenohipófisis a excepción de PRL, que asciende. Sección del Tallo Hipofisiario RESUMEN Los tumores hipofisiarios producen efectos de masa y efectos endocrinos Saber estudiar el hipopituitarismo inducido por tumores Diagnóstico de acromegalia Diagnóstico de prolactinoma Diagnóstico diferencial de aumento de volumen selar
- 17. HIPOPITUITARISMO 1)Etiología 2)Cuadro Clínico: -comienzo insidioso -pérdida progresiva de ejes 3)Diagnóstico: clínico-laboratorio- radiológico 4)Tratamiento HIPOPITUITARISMO.CAUSAS 1)Déficit aislado de ejes: congénito o adquirido 2)Tumores: macroadenomas hipofisiarios apoplejia hipofisiaria T. hipot-hipof.: craniofaringiomas 3)Inflamación: linfocitis hipofisiaria 4)Vascular: síndrome de Sheehan aneurisma de la carótida 5)Traumática: cirugía sección tallo hipot-hipófisiario HIPOPITUITARISMO. CUADRO CLINICO Déficit de GNS: -amenorrea e infertilidad - líbido e impotencia - de la barba y rasaje Déficit de GH: -retardo de crec. en niños -adultos: fatiga-repartición androide de grasa, arrugas finas faciales Déficit de PRL: -agalactia HIPOPITUITARISMO. CUADRO CLINICO Déficit de TSH: hipotiroidismo Déficit de ACTH: fatigabilidad-anorexia- de peso- pigmentación piel y areóla vello axilar y pubiano- hipoglicemia (en niños asociado a déf. GH) Déficit de ADH: menos frecuente: sindrome poliurio-polidipsia
- 18. HIPOPITUITARISMO. LABORATORIO 1) exs. inespecíficos: anemia normo-normo- hipoglicemia 2) exs. específicos: cortisol bajo y no estimulable (hipoglicemia insulínica-metopirona)- GH no estimulable (hipoglicemia-arginina- clonidina) GNS que no responden a GN-RH, estradiol con FSH normal, testosterona con LH normal T3-T4 bajas con TSH normal HIPOPITUITARISMO. TRATAMIENTO EJE SUPRARRENAL: cortisol: 20 mg/día o prednisona: 5 mg/día EJE TIROIDEO:100 ug/día EJE GONADAL: estrógenos-progesterona testosterona eje hormona de crecimiento: imprescindible en el niño Síndrome Sheehan Síndrome de la silla turca vacía Cuando la hipófisis no llena la silla turca , el espacio restante es ocupado por LCR Se puede presentar de dos formas: Primaria: no hay evidencia de tumor preexistente. Clasicamente se asocio a personas obesas, multiparas e hipertensas se asocia a un aumento de presión del LCR . Función hipofisiaria normal, pero puede haber PRL alto. Si desplaza quiasma óptico o produce rinorrea es de tto quirúrgico
- 19. Silla turca vacía secundaria Se produce después de un infarto o destrucción (cirugía, rt) de una hipófisis aumentada de tamaño o portadora de un adenoma. Se reponen las hormonas deficitarias PATOLOGIA DE LA GLANDULA SUPRARRENAL ESTRUCTURA: Forma variable, plegadas irregularmente y aplanadas. Tamaño: 5x2x1cm Peso: 7 - 10 gramos Superficie de sección : corteza amarillenta con fina banda grisácea profunda que la separa de una medula interna de color marrón CORTEZA SUPRARRENAL ESTRUCTURA Cada glándula suprarrenal está formada por una zona interna denominada médula y una zona externa que recibe el nombre de corteza. Las dos glándulas se localizan sobre los riñones. Corteza: - Zona glomerular (10%)-mineralc. - Zona Fasciculada (80%)-cortic.hsexu - Zona reticular (10%)-cortic.hsexuales CORTEZA SUPRARRENAL
- 20. REGULACION DE LA SINTESIS La biosíntesis de glucocorticoides esta regulado por un feed-back negativo con hipófisis, por el ritmo circadiano, y por la secreción de CRH estrés- dependiente. El ritmo circadiano determina un pico matinal, una disminución hacia medio día y un nadir nocturno. Su inactivación se produce a nivel hepático por reducción de uno de los anillos y luego se excretan por vía renal. EFECTOS DEL CORTISOL Carbohidratos: Aumento de la glicemia, por antagonismo y supresión de la insulina. Estímulo a la gluconeogénesis. Proteínas: Aumento del catabolismo proteico y excreción de nitrógeno. Inhibición de la síntesis proteica y movilización de aminoácidos para la gluconeogénesis. Grasas: Activación de la lipasa, aumento de la movilización de ácidos grasos. EFECTOS DEL CORTISOL Acción antiinflamatoria, que se ejerce en la microvasculatura y en diversas líneas celulares. En la microvasculatura disminuyen la permeabilidad capilar y facilitan la vasoconstricción de arteriolas. Aumentan los polimorfonucleares, disminuyen la actividad de linfocitos T y reducen los eosinofilos, alterando la inmunidad celular y humoral. Inhiben la producción y/o acción de mediadores locales de inflamación (linfokinas y prostaglandinas) LIVER GLUCONEOGENESIS ENZYME SYNTHESIS GLUCOSE GLYCOGEN (RELATIVE?) GLUCOSE CONNECTIVE SKIN ADIPOSE LYMPHOID MUSCLE AMINO ACIDS GLYCEROL FATTY ACIDS ENERGY SUPPLY
- 21. CUSHING EFECTOS METABOLICOS DEL HIPERCORTISOLISMO Aumento y redistribución de la grasa corporal (obesidad central, facies de luna). Aumento del catabolismo proteico (emaciación, osteoporosis). Aumento de la gluconeogénesis y de la resistencia insulinica (diabetes mellitus). Involución del tejido linfático y disminución de la respuesta inflamatoria (susceptibilidad a infecciones). Retención de sodio y agua (hipertensión arterial). EFECTOS METABOLICOS DEL HIPERCORTISOLISMO Obesidad centrípeta 80-90% Debilidad Muscular: 50-90% Estrías : 50-60% Hirsutismo: 65-75% Fragilidad capilar: 35-75% Hipertensión arterial: 65-75% Trastornos menstruales: 60-80% Trastornos mentales: 60-80% Diabetes Mellitus: 20-40%
- 22. CAUSAS DEL EXCESO DE CORTISOL Dependiente de ACTH: Independiente de ACTH: Enfermedad de Cushing ACTH ectópico CRH ectópico Tumor suprarrenal(adenoma y carcinoma) Hiperplasia micronodular y macronodular Fármacos (ej. corticoides) ¿Qué es lo primero para diagnosticar Síndrome de cushing? CUSHING Excreción urinaria de cortisol en 24 horas: es la forma más práctica más directa y más especifica para conocer la producción de cortisol y es la primera prueba que tenemos que realizar Si el valor obtenido es 3 veces superior al limite máximo se hace el diagnostico definitivo de s cushing. Valores positivos: 100-140 ug/dia Screning Otro test de screning podría ser el test de 1 mg de Dexametasona o test de Nugent . Se administra un mg de dexametasona a las 11pm o 12 pm y se realiza una determinación del cortisol a la mañana siguiente en ayunas . La falta de supresión por debajo de un límite (clásicamente 5ug/dl, actualmente <1.8 ug/dl) hace esta prueba positiva
- 23. Para establecer diagnostico definitivo Test clásico de 2mg de dexametasona Little débil Administrando 0.5 mg /6h durante 48 horas , es decir 2mg durante 2 días. Se hace el diagnostico definitivo de síndrome de Cushing cuando el cortisol plasmático no suprime adecuadamente Una prueba adicional: cortisol nocturno: fisiológicamente el cortisol entre 23- 24 horas está suprimido. La comprobación de este hecho descarta S de Cushing y la falta de supresión lo diagnosticaría ACTH Si ACTH es < 10 con RIA (radioinmunoensayo) o <5pg/ml con IRMA (método inmunoradiométrico), más sensible) es ACTH independiente y directamente se procede a realizar una prueba de imagen suprarrenal. Luego determine SINDROME DE CUSHING (ACTH INDEPENDIENTE)
- 24. Adenoma benigno adrenal ectópico Adenoma Cortical Tumor nodular bien circunscrito. Generalmente < de 5cm. Encapsulado y bien delimitado. Zona de fibrosis, hemorragia. Corteza adyacente atrofica Adenoma Cortical: Síndrome de Cushing Histología: Células grandes, uniforme dispuesta en nidos y trabéculas. Abundantes citoplasma, llenas de lípidos. Núcleos pequeños, regulares. NO TODOS LOS ADENOMAS PRODUCEN HORMONAS. (no funcionantes 5%).
- 25. Cancer adrenal Típicamente adenomas adrenales son más pequeños que los carcinomas. La presencia de calcificación , hemorragia y necrosis orienta hacia el diagnóstico de carcinoma CARCINOMA SUPRARENAL La evidencia de virilizacion (hirsutismo, clitoromegalia, calvicie) por producción de andrógenos. En el varón si es productor de estrógenos produce ginecomastia y en la mujer hemorragias disfuncionales HIPERPLASIA ADRENAL- TAC
- 26. HIPERPLASIA ADRENAL BILATERAL -RMN adenoma carcinoma hiperplasia DHEA-s normal Elevado (virilizante) normales imagen >2cm =TAC >6cm TAC Tac o RM bilateral tratamiento Suprarrenal ectomia uni Idem masa gangios Suprarenalec tomia bilat SINDROME DE CUSHING ACTH DEPENDIENTE ACTH DEPENDIENTE El estadio final diagnóstico es determinar la fuente de secreción de ACTH La mayoría de pacientes tienen adenoma pituitario productor de ACTH Deben realizarsele test no invasivos (test de dexametasona a dosis altas y el test de estimulación con CRH. La supresión de cortisol después de la administración de dexametasona y el incremento de ACTH y cortisol después de la administración de CRH hace el diagnostico de adenoma hipofisiario (enfermedad de Cushing)
- 27. TEST DE DEXAMETASONA ALTAS DOSIS(test de liddle fuerte) Se dá dexametasona 8 mg típicamente a las 11pm y se toma una muestra de cortisol 8am Cortisol < de 5 ug/dl o < 140 nmol/l aparece en pacientes con enfermedad de Cushing (microadenoma hipofisiario)- En pacientes normales los niveles son indetectables Una estrategia alternativa es si se compara el valor de cortisol tomado después de la prueba con un cortisol basal y si se encuentra una disminución > a 90% el diagnostico está hecho. Se ha establecido que los macroadenomas hipofisiarios tampoco suprimen con el test TEST DE DEXAMETASONA ALTAS DOSIS Tiene la ventaja de que la secreción de ACTH por adenomas hipofisiarios es relativamente resistente(y por lo tanto solo responden a dosis altas)a la regulación feedback negativa de los corticosteroides a diferencia de los tumores no hipofisiarios secretores de ACTH que son completamente resistentes al la inhibición por feedback (excepto algunos carcinoides bronquiales- raro) En Las neoplasias suprarrenales tampoco se produce supresión TEST DE CRH La mayoría de pacientes con enfermedad de Cushing responden con mayor respuesta de ACTH y cortisol luego de la administración de CRH. Pacientes con tumores suprarrenales con cortisol libre urinario incrementado y la mayoría de pacientes con secreción de ACTH ectópica no responden porque la secreción de ACTH hipofisiaria está suprimida
- 28. Estudios Imagenológicos Sirve para localización tumoral La RMN es más sensitiva que la TAC en detectar adenomas pero aún así solo detecta el 50% de esos tumores- RMN dinámica después de la administración de gadolinio es más especifica No se usa para detectar tumores ectópicos secretores de ACTH ENFERMEDAD DE CUSHING: RMN SINDROME DE CUSHING ECTOPICO Surge de la producción autónoms de ACTH o CRH a partir de enfermedades tumorales extrahipofisiarias . Tumores carcinoides (pulmón, timo, intestino, páncreas, ovario), carcinoma microcitico de pulmón (hasta el 50% de los casos) tumores de islotes pancreáticos, carcinoma medular de tiroides, feocromocitoma, paragangliomas Cateterismo de senos petrosos inferiores Los microadenomas hipofisiarios pequeños que no se visualizan en la RM se debe realizar un cateterismo bilateral de senos petrosos inferiores La demostración de un gradiente petroso periférico de ACTH (mayor nivel en seno petroso que en vena periférica) permite localizar el lugar de hipersecreción de ACTH en la hipófisis. Si no hay gradiente el dx es SC ectópico productor de ACTH
- 29. SOSPECHA DE CUSHING Cortisol libre urinario Prueba de Nugent Normal Dudoso Anormal No es Cushing Posible Cushing Cortisol plasmático 23:00 h Prueba de Liddle dosis baja Prueba de Liddle dosis alta Normales No es Cushing Anormales Cushing TRATAMIENTO Síndrome de Cushing: el tratamiento de las enfermedades adrenales primarias involucra la extirpación de estas. Los tumores adrenales son removidos con adrenelectomia unilateral mientras adrenelectomia bilateral se utiliza para pacientes con hiperpalsia bilateral macro o micronodualr TRATAMIENTO Los adenomas son siempre curados con adrenelectomia unilateral a diferencias el carcinoma donde fácilmente recurren e incluso aún con radioterapia y quimioterapia. Única opción: mitotano TRATAMIENTO ENFERMEDAD DE CUSHING Cirugía: hipofisiaria (generalmente transesfenoidal) o suprarrenal= microadenectomía. Hipofisectomia subtotal si microadenoma no es encontrado y/o no hay problemas de fertilidad En Cushing ectópico cirugía es del tumor productor de ACTH. Radioterapia: hipófisis en los que no se puede hacer cirugía en las recaídas post quirúrgicas. Y es terapia de inicio en < 18(acelerador linel-85% de respuesta) Terapia medicamentosa (metopirona, aminoglutetimida, ketoconazol o mitotane) Cura final: bilateral adrenelectomia con reemplazo glucocortic y mineralc. es la cura definitiva.
- 30. Transsphenoidal surgery 75-90 percent initial cure 5-20 percent recurrence 60-70 percent final cure 75-90 percent initial cure 20-40 percent recurrence 45-50 percent final cure Pituitary irradiation mitotane Total bilateral adrenalectomy 85-100 percent cure 100 percent cure Adults Children Metirapona o Metopirona -Interfiere con la síntesis de cortisol y de corticosterona -Origina desvanecimientos transitorios y trastornos gastrointestinales -En dosis de 0.25g/12horas a 1g/6horas puede reducir la producción de cortisol a concentraciones normales en pacientes con tumores suprarrenales, síndrome de ACTH e hiperplasia. -Se utiliza con mayor frecuencia en pruebas de funcionamiento suprarrenal Aminoglutetimida Bloquea la conversión del colesterol a pregnenolona y reduce la síntesis de todos los esteroides hormonalmente activos. Se usa para reducir o eliminar la producción de estrógeno y andrógeno en pacientes con carcinoma de mama. Se puede usar junto con ketoconazol para reducir la secreción de esteroides en pacientes con síndrome de Cushing debido a cáncer adrenocortical que no responde al mitotano. Cholesterol Pregnenolone Progesterone 17-hydroxypregnenolone 17-hydroxyprogesterone DHEA Androstenedione Deoxycorticosterone Corticosterone Aldosterone Cortisol 11-deoxycortisol Testosterone Dihydrostestosterone DHEAS SL Estrone Estradiol 17,20 17 17 17,20 3 3 3 17 R 17,20 21 11 18 11 21 5 R 17 R A A SK
- 31. KETOCONAZOL Es un antimicótico derivado del imidazol Inhibe la síntesis de esteroides suprarrenales y gonadales Inhibe la fragmentación de las cadenas colaterales del colesterol y a las enzimas que se requieren para síntesis de glucocorticoides, estos efectos son menores en enzimas de mamíferos que en enzimas micoticas por lo que se necesitan dosis altas para ver efectos. Se ha utilizado para la terapéutica de pacientes con síndrome de cushing con dosis de 200 a 1200 mg/día han reducido las concentraciones de hormonas Mitotane Ocasiona atrofia suprarrenal en perros e interfiere con las vías biosintéticas en la corteza suprarrenal. Se administra por vía oral en dosis divididas de 6 a 12 mg/día 1/3 de pacientes con carcinoma adrenal mostraron disminución en la masa tumoral y la producción de esteroides fue disminuida en dos tercios Efectos como diarrea, nauseas, vomito, depresión, somnolencia y trastornos cutáneos. Fue retirado en los EUA Mifepristona (RU 486 Ocasiona resistencia glucocorticoide generalizada Actualmente solo se recomienda para pacientes inoperables con secreción ectopica de ACTH o carcinoma suprarrenal que fallaron a otras medidas terapéuticas ALTERACIONES DE LA CORTEZA SUPRARRENAL SINDROME ADRENOCORTICAL (virilismo suprarrenal). Causas: - Carcinoma suprarrenal secretor de andrógenos. - Hiperplasia suprarrenal congénita por déficit hereditario que interviene en la biosíntesis del cortisol.
- 32. ALTERACIONES DE LA CORTEZA SUPRARRENAL SINDROME ADRENOCORTICAL Cuadro clínico: Desarrollo precoz del niño, pubertad precoz Virilización en niños (déficit completo) Alteraciones electrolíticas ( gran eliminación de orina que origina hipotensión debido a insuficiencia de cortisol y aldosterona. ALTERACIONES DE LA CORTEZA SUPRARRENAL SINDROME ADRENOCORTICAL Asociada a déficit enzimático de hiposecreción y del tipo de hipersecreción inducida por el bloqueo Déficit completo de 21-hidroxilasa (30%) Déficit parcial del 21-hidroxilasa (60%) Déficit de 11-hidroxilasa (5%) Otros: 17-hidroxilasa. Cholesterol Pregnenolone Progesterone 17-hydroxypregnenolone 17-hydroxyprogesterone DHEA Androstenedione Deoxycorticosterone Corticosterone Aldosterone Cortisol 11-deoxycortisol Testosterone Dihydrostestosterone DHEAS SL Estrone Estradiol 17,20 17 17 17,20 3 3 3 17 R 17,20 21 11 18 11 21 5 R 17 R A A SK Ambas glándulas aumentan de tamaño y peso Engrosamiento difuso o nodular Microscópicamente la zona fasciculada y la reticular son mas anchas de lo habitual. Las células se parecen a las normales. HIPERPLASIA CORTICAL BILATERAL
- 33. Deficit clasico de 21 hidroxilasa Forma compensada: virilizante , cortisol normal y aldosterona normal. Forma grave: deficit de mineralcorticoides y glucocorticoides Es la causa más frecuente de genitales ambiguos en el RN. Mujer: desde el nacimiento: hipertrofia de clitoris, fusión labioescrotal, virilizacion de la uretra. Los genitales internos son femeninos con involución de los conductos de Wolf, gonadas normales. Si no setrata las mujeres sufren virilizacion progresiva , talla corta Deficit clasico de 21 hidroxilasa En los varones este defecto se diagnostica hasta los 2- 3años con crecimiento acelerado, maduración de los genitales externos, cambio de voz, maduración de caracteres sexuales secundarios. Si no se tratan :talla corta Principal esteroide secretado: 17- hidroxiprogesterona Déficit de 11 hidroxilasa Cortisol y aldosterona disminuidas Grado de virilizacion: igual que 21. Genitales internos y gonadas normales Se produce HTA por aumento de la DOCA , un precursor de aldosterona con potente efecto de retención de sal Principal esteroide secretado: 11 desoxicortisol y desoxicorticosterona Déficit de 3 beta OH esteroide deshidrogenasa Déficit de cortisol y aldosterona. La síntesis de andrógenos es detenida en la dehidroepiandrosterona que es un andrógeno débil virilización leve e la mujer, ausencia de virilización en el hombre (DHEA es andrógeno débil)
- 34. INSUFICIENCIA SUPRARENAL DEFICIT DE CORTISOL: CAUSAS Primarias: (Enf. de Addison) Destrucción de la glándula (autoinmunidad, TBC) Resección de ambas suprarrenales. Secundarias: (por falla hipofisiaria) Destrucción de la adenohipófisis (Tumores, Sheehan). Iatrogénica (cirugía o radioterapia). EFECTOS METABOLICOS DEL HIPOCORTISOLISMO: CUADRO CLINICO Síntomas generales: debilidad, fatiga, vómitos, adelgazamiento. Hipotensión arterial: por disminución de la retención de sodio-agua y menor reactividad vascular. Hiperpigmentacion en los casos de falla adrenal primaria debido al aumento de ACTH y de la hormona melanocito estimulante. Apetencia por la sal. Hiponatremia e hiperkalemia (si se asocia un déficit de mineralocorticoides). ENFERMEDAD DE ADDISON Requiere destrucción > 90 % de la glándula 50 % tienen Acs. antiadrenales circulantes Otro % tienen Acs contra otras glándulas endocrinas (tiroides, paratiroides, páncreas, gónadas)
- 35. SINTOMAS SIGNOS/SINTOMAS % fatigabilidad 99 hiperpigmentación cutánea 98 pigmentación de mucosas 82 baja de peso 97 anorexia, náuseas, vómitos 90 hipotensión (110/70) 87 SIGNOS Y SINTOMAS SIGNOS Y SINTOMAS dolor abdominal 34 % avidez por la sal 22 % diarrea 20 % constipación 19 % síncope 16 % vitiligo 9 % otra manifestaciones: cambios marcados de personalidad, del sentido gusto-olfato-audición, vello axilar y pubiano en mujer Hiperpigmentación Hiperpigmentación
- 36. LABORATORIO 1) Exámenes inespecíficos: hiponatremia-hipocloremia, hiperkalemia, acidosis metab., hipercalcemia leve a moderada, anemia normo. normocrómica, linfocitosis relativa, eosinofilia moderada Rx EEG: de ondas y enlentecimiento generalizado Corazón en gota LABORATORIO 2) Pruebas Específicas: prueba de ACTH: medición basal de cortisol plasm. y 30- -inyección de (ACTH): respuesta normal: cortisol > 18 ug/dl o diferencia de > 7 ug/dl déficit severo : cortisol 8 AM < 2ug/dl LABORATORIO Disminución del cortisol en sangre y orina Ausencia de elevación del cortisol al estimulo agudo con ACTH. Elevación de ACTH en los casos de Enfermedad de Addison y disminución en los casos de enfermedad hipofisiaria. Determinación de anticuerpos anti-corteza suprarrenal o hipófisis. Estudio de imágenes (tu. Hipofisiario, suprarrenal).
- 37. DIAGNOSTICO DIFERENCIAL Síndrome depresivo: fatigabilidad marcada con aumento de peso Hipotiroidismo: fatigabilidad con aumento leve de peso y otros síntomas propios TRATAMIENTO Reemplazo de glucocorticoides y mineralocorticoides: Usar solo cortisol (Enf. hipofisiaria) o asociado a fludrocortisona (Enf. Suprarrenal) En situaciones de estrés la dosis de cortisol debe ser aumentada. Tratamiento etiológico cuando se justifique (TBC, Sarcoidosis, etc). TRATAMIENTO Educación, debe llevar información reemplazo de GC: cortisol ( 20 mg (8 AM, 10 mg 16 PM) reemplazo de MC: fludrocortisona (0,05-0,1 mg) CRISIS SUPRARRENAL AGUDA Definición: cuadro agudo de carencia de GC y MC con riesgo vital Causas: a) Insuf. SR Crónica preexistente AGRAVADA o DESCOMPENSADA por stress agudo (sepsis- cirugía-trauma) b) Suspensión brusca de GC exógenos Cuadro Clínico: intensificación de síntomas: pseudoabdómen agudo-vómitos profusos- compromiso de conciencia-shock-muerte
- 38. CRISIS SUPRARRENAL AGUDA: TRATAMIENTO Hospitalización en UCI-UTI Reposición de LEC y Na Reemplazo de GC: 100 mg IV de hidrocortisona en bolo, seguido de hidrocortisona 10 mg/hr Uso de drogas vasoactivas Endocrinologia Respecto al, síndrome de hiperfunción adrenal (síndrome de Cushing) Señale lo incorrecto: 1. La piel es engrosada y pálida 2. Cursa con hipertensión arterial 3. Presencia de estrías rojo- purpúricas superficiales 4. Hirsutismo 5. Obesidad centripeta ENFERMEDAD DE CUSHING Por exceso de glucocorticoides, incluye tipo iatrogénico. Piel delgada y atrófica Reparación de heridas es inhibida Estrías grandes y color púrpura Apariencia pletórica y predominan Telangiectasis ENFERMEDAD DE CUSHING Fascie abotagada, aumento del tejido celular graso en espalda y cuello (giba de búfalo) y obesidad troncal. Los cambios cutáneos son reversibles parcialmente: estrías y telangiectasias pueden aclarse pero no desaparecen.
- 39. ESTRIAS Las estrias aparecen debido a que la piel al ser delgada no puede esconder el color de los vasos sanguíneos en la dermis subyacente. Usualmente es en abdomen y tronco pero tam´bién se le puede ver en mamas, brazos piernas, mamas y hombros.
- 40. Equimosis Perdida de tejido conectivo subcutaneo por efecto catabólico del corticoide resulta en sangrados despues de un trauma menor. RESPUESTA: 1. la piel es engrosada y pálida ENDOCRINOLOGIA En la insuficiencia suprarrenal se presenta: 1) Hipernatremia 2) Hipocalemia 3) Hipoglicemia 4) Hipocalcemia 5) Alcalosis metabólica INSUFICIENCIA SUPRARENAL SINTOMA FRECUENCIA Debilidad, fatiga 100% Anorexia 100% Sintomas gastorintest 96% Nauseas 82% vomitos 75% Constipacion 33% Dolor abdominal 31% Diarrea 16% Avidez por sal 16% Hipot. Postural 12% Dolor musc.articular 6-13%
- 41. INSUFICIENCIA SUPRARENAL SIGNO PORCENTAJE Perdida de peso 100% Hiperpigmentacion 94% Hipotension<110 88-94% Vitiligo 10-20% Calcificacion auricular 5% INSUFICIENCIA NSUPRARENAL Anormalidades laboratorio Porcentaje Disturbios electrolitos 92% Hiponatremia-hipoglic(2) 88% Hiperkalemia 64% Hipercalcemia 6% Azoemia 55% Anemia 40% eosinofilia 17% RESPUESTA: 3).- hipoglicemia .- La insuficiencia suprarrenal primaria autoinmune cursa con: a) ACTH y cortisol no dosificables b) ACTH elevada, cortisol disminuido c) Renina elevada, aldosterona disminuida d) A y C son correctas e) B y C son correctas
- 42. La insuficiencia primaria autoinmune afecta tanto la corteza como la médula suprarrenal, por infiltración linfocítica. El cortisol y la aldosterona estarán disminuidos. Si el cortisol está bajo, la hipófisis intentará compensar la situación con la liberación de ACTH; asimismo, en el riñón un déficit de aldosterona se intentará compensar con una renina alta. ALDOSTERONA La aldosterona es sintetizada en la zona glomerulosa de la corteza suprarrenal. La biosíntesis es estimulada directamente por angiotensina II, ACTH y el potasio plasmático. El principal regulador es la angiotensina II, la cual responde a los cambios del volumen plasmático a través de la activación del SRAA. HIPERSECRECION DE HORMONAS CORTICALES HIPERALDOSTERONISMO PRIMARIO : Síndrome de Conn Hipersecreción de aldosterona. Poco frecuente. Causas: - Adenoma cortical secretor de aldosterona (70%) - Hiperplasia de zona glomerular (30%) Los carcinoma raramente segregan aldosterona. HIPERSECRECION DE HORMONAS CORTICALES HIPERALDOSTERONISMO PRIMARIO : Síndrome de Conn Clínica: Hipertensión Hipopotasemia causante de debilidad muscular y alcalosis. HIPERALDOSTERONISMO SECUNDARIO: Inducida por producción excesiva de renina angiotensina. (isquemia renal, diabetes insípida)
- 43. CUADRO CLINICO AUMENTA REABSORCIÓN DE SODIO HIPERNATREMIA HIPOKALIEMIA INTERCAMBIO CELULAR CON H+ ACIDOSIS METABOLICA HIPEROSMOLARIDAD RETENCION DE AGUA AUMENTO DEL VOLUMEN PLASMATICO HTA TRASTORNOS DEL RITMO CARDIACO TETANIA EVALUACION La certificación del HAP generalmente se realiza a través de un test de sobrecarga salina o de fludrocortisona (mineralocorticoide sintético). En ambas pruebas se persigue suprimir la producción de aldosterona (generalmente a <5ng/dl), lo cual de no lograrse certifica su producción autónoma y con ello él diagnostico de HAP La relación AP/ARP > 25 es muy sugestiva y > 50 casi diagnóstica HIPERTENSOS ESENCIALES Relación AP/ARP >25 Test de Fludrocortisona Hiperaldosteronismo 1º Test de Dexametasona Hiperplasia Adenoma Cirugía Espironolactona Fardella C, Mosso L, Montero J et al. J Clin Endocrinol Metab (2000) 85:1863-1867. Ap = Aldosterona plasmática ARP = Actividad de renina Plasmatica ANTAGONISTAS MINERALOCORTICOIDES Compiten con la aldosterona por los sitios de fijación y disminuyen su efecto periféricamente Espironolactona 7- -acetiltioespironolactona se usa en el tratamiento del hiperaldosteronismo primario en dosis de 50 a 100 mg/día.
- 44. ANTAGONISTAS MINERALOCORTICOIDES Esplerenona Antagonista de aldosterona nuevo el cual se encuentra bajo estudios clínicos avanzados Drospirenoma Es una progestina Es un anticonceptivo oral reciente Además antagoniza los efectos de la aldosterona MEDULA SUPRARRENAL Deriva de la cresta neural Comprende al 10% del peso de la glándula. Están en contacto con terminaciones nerviosas y forma parte del sistema nervioso simpático, asumiendo el papel de 2da. Neurona vegetativa. La secreción de adrenalina y Noradrenalina esta bajo el control de los nervios simpáticos. TUMORES DE LA MEDULA SUPRARRENAL FEOCROMOCITOMA GANGLIONEUROBLASTOMA NEUROBLASTOMA Tumor productor de catecolaminas Poco frecuente. Algunos presentan HC familiar. - Herencia autosómica dominante - Asociado a neurofibromatosis: - Forma parte del síndrome MEN IIa y IIb Frecuente en adulto ( 40 - 60 años) , M>H El feocromocitoma es una causa infrecuente de HTA secundaria (0.1% de la población de hipertensos). Se origina en una hipersecreción de catecolaminas generalmente secundaria a un tumor localizado en la medula suprarrenal. Es importante su diagnostico y tratamiento oportuno, ya que tiene una alta tasa de morbi-mortalidad. FEOCROMOCITOMA
- 45. Tumor del 10% 10% Se da fuera de la glándula suprarrenal. 10% Son tumores múltiples. Afectan ambas glándulas, pero también a otros paraganglios. 10% Muestran signos de malignidad. En el sentido de crecimiento invasivo local y metástasis. 10% Aparece en niños. FEOCROMOCITOMA CUADRO CLINICO Hipertensión arterial mantenida y/o en crisis. Las crisis hipertensivas se pueden manifestar por una triada clásica: cefalea, palpitaciones y sudoración. Hipotensión ortostática por disminución de la volemia. Cambios de carácter, baja de peso e intolerancia al calor. DIAGNOSTICO Elevación de catecolaminas y metanefrinas urinarias. Confirman Dx en 95% de casos Elevación de catecolaminas plasmáticas, especialmente en relación a las crisis. Se realizan para confirmar el diagnóstico. Su medición debe realizarse después de 30' de reposo en posición supina. Un valor de 2000 pg/ml confirma el diagnóstico Estudios de localización: Resonancia nuclear magnética, tomografía computada, cintigrafía con MIBG, Octeotride. Pruebas funcionales (ej. Clonidina, Glucagón). DIAGNOSTICO Test de clonidina: no debe realizarse en pacientes hipovolémicos por el riesgo de hipotensión, ni en pacientes con catecolaminas plasmáticas normales. El test consiste en la administración oral de 0.3 mg de clonidina. Las catecolaminas plasmáticas deben medirse antes y 3hs después de la clonidina. La caída de las catecolaminas por debajo de 500 pg/ml brinda un 90% de diagnóstico de feocromocitoma.
- 46. DIAGNOSTICO Test de glucagón: por el riesgo de hipertensión, los pacientes pueden ser previamente tratados con alfa bloqueantes o bloqueantes de canales del calcio. Consiste en la administración de 1 mg de glucagón endovenoso y se realiza un dosaje de catecolaminas basal y 2 minutos después de la infusión del glucagón. El incremento de 3 veces el valor basal es diagnóstico de feocromocitoma TRATAMIENTO El tratamiento siempre será quirúrgico, luego de una adecuada preparación medica, con una curación de la HTA, después de la exéresis del tumor que oscila entre 50-70%. Tratamiento médico: se deben comenzar con Alfa- bloqueantes: doxazosin, prazosin o fenoxibenzamina en dosis crecientes y luego agregar B-bloqueantes: atenolol. También pueden utilizarse alfa-beta bloqueantes: labetalol, y hay buenos resultados con bloqueantes del canales del calcio. Las crisis hipertensivas se tratan con fentolamina endovenosa 2-5 mg cada 5 minutos hasta controlar la TA, o con nitroprusiato de sodio. NEUROBLASTOMA Tumor maligno originado en la cresta neural. Frecuente en niños. Edad media 2 años. El 80% <de los 5 años. Localización frecuente: Medula suprarrenal. Extra abdominal: Mediastino posterior, pelvis, cabeza, cuello, neuroblastoma olfatorio (individuos mayores). NEUROBLASTOMA Característica citogénetica: Delección parcial (monosomía parcial) brazo corto del cromosoma 1 Evolución: Metástasis precoz (hígado, órbita, ósea) Cirugía, RT,QT. Pronostico: Depende de la edad. Estadio I : Limitado al órgano Estadio II : Diseminación local unilateral Estadio III : Diseminación local, compromiso ganglios regional. Estadio IV : Metástasis a distancia.
- 47. NEOPLASIA ENDOCRINAS MULTIPLES (NEM) Síndrome caracterizado por la aparición sincrónica o asincrónica de varios tumores y/o hiperplasia de glándulas endocrina. Se puede dar en forma esporádica o de carácter familiar con herencia autosómica dominate. Son multicéntricos. NEOPLASIA ENDOCRINAS MULTIPLES (NEM) Tipos: NEM I : Síndrome de Wermer NEM II a : Síndrome de Sipple NEM II b NEOPLASIA ENDOCRINAS MULTIPLES I (Sínd. Wermer) Mutación alélica heredada de carácter recesivo en el cromosoma 11 (11q13) Se presenta con tumores o hiperplasias de: - Adenohipófisis - Paratiroides - Corteza suprarrenal - Páncreas - tiroides Junto con ulcera péptica e hipersecreción gástrica. Síndrome de Zollinger - Ellison (Adenoma insular pancreático asociado a úlcera péptica) NEOPLASIA ENDOCRINAS MULTIPLES IIa (Sínd. Sipple) Presenta una combinación de: - Feocromocitomas - Carcinoma medular de tiroides - Hiperplasia o adenoma de paratiroides No aparecen neuromas mucocutáneos
- 48. NEOPLASIA ENDOCRINAS MULTIPLES II b Cursa con neuromas mucocutáneos en: - Párpados, lengua, labios, intestinos, bronquios y vejiga. El gen de los síndromes NEM IIa y IIb: Se deben a una mutación del protooncogén ret (10q11.2). - En los axones 10 ó 11 el IIa. - En los axones 16 el IIb. NEOPLASIA ENDOCRINAS MULTIPLES Manifestaciones clínicas son variadas. Depende de la lesión funcionante. Su estudio se puede determinar por técnicas de biología molecular, facilitando el diagnóstico de portadores mediante rastreo familiar. ENDOCRINOLOGIA Se habla de malignidad en un feocromocitoma: 1. Nunca son malignos. 2. Cuando invaden localmente o metastatizan. 3. Cuando tienen más de 6 cm de diámetro. 4. Cuando aparecen atipias y otros signos histológicos de malignidad. 5. Cuando secretan dopamina.
- 49. FEOCROMOCITOMAS Son tumores que sintetizan y liberan catecolaminas poco frecuentes habitualmente benignos que proceden de las células cromafines del sistema nervioso simpático (medula suprarenal y ganglios simpáticos paraganglionares) La mayoría aparece entre la 4ta y 5ta. Década de la vida, la mayoría son mujeres RESPUESTA: 2.- cuando invaden localmente o metastatizan FEOCROMOCITOMAS El 80% son lesiones únicas y unilaterales el 10 % son bilaterales y el 10% están fuera de las g. Suprarenales. Menos del 10% sigue un curso evolutivo maligno y su malignidad como pasa en otros tumores endocrinos no depende del estadio histológico sino de la invasión local y de la presencia de metástasis ENDOCRINOLOGIA - Para descartar feocromocitoma, solicitaría inicialmente: 1. Catecolaminas en plasma. 2. Prueba con glucagón. 3. Prueba con fentolamina. 4. Catecolaminas libres y metanefrinas en orina de 24 h. 5. Gammagrafíacon metayodobencilguanidina - I- 131.
- 50. Pruebas diagnosticas en el feocromocitoma PRUEBA SENSIBILIDAD ESPECIFICIDAD Catecol. orina 100% 98% Metane.orina 91% 100% TAC abdominal 90-98% 70% RM abdominal 98-100% 70% Gama 90% 100% RESPUESTA: 4.- catecolaminas libres y metanefrinas en orina de 24 horas 46.- Paciente varón de 27 años de edad, que tras ser diagnosticado hace 2 meses de un Feocromocitoma, se le descubre un nódulo tiroideo frío en una gammagrafía tiroidea. En la analítica resalta un aumento desproporcionado de calcitonina plasmática. ¿En que patología habría que pensar? a) Adenoma tiroideo b) Carcinoma medular de tiroides c) Carcinoma papilar de tiroides d) Carcinoma folicular de tiroides e) Carcinoma paratiroieo Ante la presencia de un nódulo tiroideo en un paciente diagnosticado de feocromocitoma es obligado descartar la existencia de un cáncer medular de tiroides, dada su asociación en el MEN 2 Además informan sobre niveles aumentados de calcitonina, que es el principal marcador tumoral del cáncer medular de tiroides. El MEN 2a asocia el cáncer medular que es la manifestación más frecuente, con feocromocitoma e hiperparatiroidismo primario por hiperplasia glandular El MEN 2b asocia el cáncer medular con el feocromocitoma, ganglioneuromatosis y hábito marfanoide.(que de diferencia del marfan típico porque no hay alteración cardiaca ni ectopia lentis)
- 51. Classification of Multiple Endocrine Neoplasia Type 1 Primary hyperparathyroidism (>90 percent) Pituitary tumors (10 to 20 percent) Prolactinoma Growth hormone-secreting Corticotropin-secreting Non-hormone-secreting Enteropancreatic tumors (60 to 70 percent) Gastrinoma (Zollinger-Ellison syndrome) Insulinoma Vasoactive-intestinal polypeptide-secreting Glucagonoma Pancreatic polypeptide-secreting Non-hormone-secreting Other Familial medullary thyroid cancer (variant of 2A) Medullary thyroid cancer Type 2A Medullary thyroid cancer (>90 percent) Pheochromocytoma (40 to 50 percent) Parathyroid hyperplasia (10 to 20 percent) Cutaneous lichen amyloidosis Type 2B Medullary thyroid cancer Pheochromocytoma Other Mucosal neuromas Intestingal ganglioneuromas Marfanoid habitus ALTERACIONES EN LAS GONADOTROFINAS Hipotálamo Hipófisis Testículo GnRH FSH Testost Estrad LH Inhibina
- 52. Gonadotropin Releasing Hormone (GnRH, LHRH) Pyro- Glu His Trp Ser Tyr Gly Leu Arg Pro Gly -NH2 Producido por neuronas del núcleo arcuato y del área preóptica (Generador de pulsos de GnRH) Mantiene la secreción gonadotrófica con la exposición de la hipófisis a la secreción pulsátil de GnRH (El rango crítico de la frecuencia en el varón es de 90 a 120 min) CLOCK TIME (h) SLEEP ADULTHOOD (Boyar et al, J Clin Invest 1974) CUADROS CLINICOS CLASIFICACIÓN 1.-HIPOGONADISMO HIPERGONADOTRÓPICO 2.-HIPOGONADISMO HIPOGONADOTRÓPICO 3.-RESISTENCIA A LA ACCIÓN DE LOS ANDRÓGENOS
- 53. HIPOGONADISMO HIPERGONADOTRÓPICO ALTERACIÓN DE CÉLULAS DE LEYDIG ANOMALÍAS DE PRODUCCIÓN DE TESTOSTERONA LESIONES DE LOS TÚBULOS SEMINÍFEROS CON OLIGOSPERMIA O AZOOSPERMIA ELEVACIÓN DE GONADOTROPINAS HIPOGONADISMO MASCULINO HIPOGONADISMO PRIMARIO Síndrome de Klinefelter (Clásico y variantes) Síndrome de Bonnevie-Ulrich Síndrome de Noonan Distrofia miotónica Síndrome de Sertoli Solo Anorquia Castración (Torsión, Cirugía, Trauma) Infecciones, Tóxicos, Radiaciones, Drogas Andropausia Hipotálamo Hipófisis Testículo GnRH FSH Testost Estrad LH Inhibina HIPOGONADISMO HIPOGONADOTRÓPICO ALTERACIONES DEL HIPOTÁLAMO E HIPÓFISIS IMPIDEN SECRECIÓN GONADOTROPINAS IMPOTENCIA , INFERTILIDAD , AMBAS
- 54. HIPOGONADISMO MASCULINO HIPOGONADISMO SECUNDARIO Sindrome de Kallmann Mutaciones de genes de GnRH y del receptor de GnRH Panhipopituitarismo Trauma, Tumor o Cirugía Hipotálamo-Hipofisaria Aracnoidocele, Quiste aracnoideo Infección, Enf. Granulomatosa/Autoinmune Hipofisaria Sindrome de Prader-Willi Pubertad retrasada Andropausia Deficiencia aislada de gonadotrofinas Hipotálamo Hipófisis Testículo GnRH FSH Testost Estrad LH Inhibina RESISTENCIAA LAACCIÓN DE LOS ANDRÓGENOS DISMINUCIÓN DE LA ESPERMATOGÉNESIS GINECOMASTIA HISTORIA CLÍNICA PUBERTAD PRESENTACIÓN CLÍNICA EDAD EN QUE SE PRESENTA LA DEFICIENCIA
- 55. Secuencia de Evenntos Puberales Edad (años) Mujeres Edad (años) Varones (Marshall W , Tanner J. 1969) HISTORIA CLÍNICA ANTECEDENTES RELEVANTES DEL HIPOGONADISMO CRIPTORQUIDIA ENFERM CRÓN ALCOHOLISMO RADIOTERAPIA TRAUMA ,TORSIÓN TESTICULAR ORQUITIS ,EXPOSICIÓN A TÓXICOS , MEDICAMENTOS EXAMEN FÍSICO GRADO DESARROLLO SEXUAL TAMAÑO TESTICULAR CARACTERISTICAS EUNUCOIDES ANOSMIA GINECOMASTIA DISMINUCIÓN DE VELLO VARICOCELE DISMORFICOS
- 56. LABORATORIO ANORMALIDADES DE LAS HORMONAS DEL EJE TESTOSTERONA FSH, LH (PULSOS CADA 90-120 MIN, SE TOMA 3 MUESTRAS CON INTERVALOS DE 20-30 MINUTOS ) EXAMEN LIQUIDO SEMINAL PROLACTINA LABORATORIO TEST DINÁMICOS : ESTIMULACIÓN CON GONADOTROPINA CORIÓNICA TEST GNRH HIPOGONADISMO HIPOGONADOTROFICO Prueba de GnRH: respuesta de LH 0 30 60 90 MINUTOS 0 10 20 30 40 LH mUI/ml GnRH 100 ug HIPOGONADISMO HIPOGONADOTROFICO Prueba de GnRH: respuesta de FSH 0 30 60 90 MINUTOS 0 10 20 FSH mUI/ml GnRH 100 ug
- 57. HIPOGONADISMO HIPOGONADOTROFICO Prueba de HCG: respuesta de testosterona 0 24 48 72 HORAS 0 5 10 15 20 T (ng/ml) hCG 5000 U IMÁGENES RMN TAC ECOGRAFÍA TESTICULAR GENÉTICOS HIPOGONADISMOS HIPERGONADOTRÓPICOS PRINCIPALES CAUSAS
- 58. DIAGNÓSTICO DIFERENCIAL KLINEFELTER: MÁS FRECUENTE PRIMARIO DISGENESIA DE TÚBULOS SEMINÍFEROS CARIOTIPO 47XXY(FORMA CLASICA) KLINELFELTER Clásicamente testículos pequeños y duros . Azoospermia, ginecomastia y aumento de los niveles de gonadotropinas en el plasma- Podría haber un mosaicismo: 46 XY/47XXY El aumento de la talla media ocurre por el predominio del segmento corporal inferior ANORQUIA BILATERAL TESTÍCULO EVANESCENTE EXISTÍAN TESTÍCULOS PERO SE REABSORBIERON ANTES O DESPUES DEL NACIMIENTO
- 59. APLASIA DE CÉLULAS LEYDIG AUSENCIA CONGÉNITA DE CEL SEUDOHERMAFRODITISMO MASCULINO ASOCIADO A AMBIGÜEDAD DE GENITALES EXTERNOS NOONAN ESPORÁDICO AUTOSÓMICO DOMINANTE HIPERELASTICIDAD PIEL HIPERTELORISMO PTOSIS OREJAS IMPLANTACIÓN BAJA NOONAN TALLA BAJA ACORTAMIENTO DE CUARTOS METACARPIANOS PALADAR OJIVAL CARDIOPATÍAS CONGÉNITAS DER COMO ESTENOSIS VAL PULM, CIA. TEST. PEQUEÑOS Y CRIPTORQ DISTROFIA MIOTÓNICA ALREDEDOR DEL 80% DE VARONES INSUFICIENCIA TESTICULAR PRIMARIA EN BIOPSIA , ALTERACIÓN ESPERMATOGÉNESIS , HIALINIZACIÓN Y FIBROSIS
- 60. TRASTORNOS DE LOS TÚBULOS SEMINIFEROS ADULTO INSUFIC IDIOPÁTICA DE TUBULOS SEMINÍFEROS O LOS QUE HAN DESARROLLADO INFECCIÓN TESTICULAR, CRIPTORQUIDIA , UREMIA , AGENTES ANTINEOPLÁSICOS , ALCOHOLISMO , RADIACIÓN,LESIÓN VASCULAR,TEMP ALTAS: OLIGOSPERMIA O AZOOSPERMIA, INFERTILIDAD HIPOGONADISMO HIPOGONADOTRÓPICO PANHIPOPITUITARISMO CONGÉNITO O ADQUIRIDO KALLMAN ANOSMIA X AGENESIA LÓBULOS OLFATORIOS E HIPOGONADISMO SECUNDARIO A DEFICIENCIA DE GnRH FALTA MIGRACIÓN NEURONAS NEUROSECRETORAS DE GnRHA DEL FETO DESDE LA PLACA OFLATORIA AL HIPOTALAMO ENFERMEDAD INFILTRATATIVA H-H HISTIOCITOSIS SARCOIDOSIS HEMOCROMATOSIS TUMORES BENIGNOS Y MALIGNOS
- 61. RETRASO CONSTITUCIONAL AUSENCIA DE DESARROLLO PUBERAL EN MENOR DE 14 AÑOS ANTECEDENTES FAMILIARES DE RETRASO DEL DESARROLLO SEXUAL EN EL PADRE O HERMANOS MAYORÍA SIGNOS PUBERALES AL LLEGAR A LOS 18 AÑOS DEFICIENCIA AISLADA LH EUNUCO FÉRTIL PERDIDA DE LH, FSH CONSERVADA PRADER WILLI OBESIDAD-HIPERFAGIA HIPOTONÍA MUSCULAR RM DIABETES DE TIPO ADULTO EN PACIENTES JOVENES TAMBIEN HAY DEFICIT DEGNRH HIPOGONADISMO HIPOGONADOTRÓFICO IDIOPÁTICO RARA MUTACION GEN GnRH
- 62. MUTACIÓN GEN SUBUNIDAD BETA LH O FSH RARO ASOCIADOS A ENF CRÓNICAS COMO: ANOREXIA, SIDA, IRC, CIRROSIS ENVEJECIMIENTO DISMINUCIÓN MÍNIMA TESTOSTERONA SÉRICA TOTAL AUMENTO DE GLOBULINA TRANSPORTADORA DE TESTOSTERONA DISMINUCIÓN DE TEST LIBRE FSH Y LH ELEVACIÓN MÍNIMA INDUCIDO POR MEDICAMENTOS ALQUILANTES: CLORAMBUCIL, CICLOFOSFAMIDA CARBOPLATINA Y CISPLATINA SURAMINA KETOCONAZOL GLUCOCORTICOIDES NEMATOCIDAS: DIBROMODICLOROPROPANO TRATAMIENTO REESTABLECER FUNCIÓN SEXUAL, LÍBIDO, SENSACIÓN BIENESTAR. PRODUCIR VIRILIZACIÓN Y MANTENERLA OPTIMIZAR DENSIDAD ÓSEA PREVENIR OSTEOPOROSIS RESTABLECER FERTILIDAD
- 63. TRATAMIENTO TESTOSTERONA IM PARCHES TRANSDERMICOS O ESCROTALES TESTOSTERONA EN GEL FSH Y LH SOSPECHA DE HIPOGONADISMO Estradiol o testosterona bajas LH y FSH Bajas Falta de inicio de pubertad con EO >12 años (mujer) o >13 años (varón) Detención de desarrollo puberal Elevadas Hipogonadismo primario Cariotipo Prueba con GnRh (100 µg) LH,FSH elevadas Hipogonadismo secundario Hipogonadismo terciario LH,FSH disminuidas Pseudohermafroditismo masculino 1.-Sintesis deficitaria de testosterona con virilizacion incompleta del embrión masculino HSClipide, déficit de 3 beta OH esteroide deshidrogenasa, déficit de 17 hidroxilasa , 17 liasa. 2.-Anomalias de la accion de androgenos- resistencia a la testosterona: femenizacion testicular o resistencia androgenica completa(Moprris)-ligada a X. NO hay órgano genital interno. Testículos en abdomen, vagina corta que acaba en fondo de saco. Mama y habito general femenino. HIPOGONADISMO
- 64. HIPOGONADISMO FEMENINO 1) Caracterizado por fallo gonadal por alteración ovárica propia o secundaria a fallo hipotalámico-hipofisiario. 2) Ocurre en distintos momentos de la vida y por causas diversas ocasionando distintas formas clínicas de presentación. 3) Ovario tiene dos funciones: produce gametos femeninos (oogénesis) y secreta hormonas esenciales para la función reproductora y regulación de la diferenciación y desarrollo de órganos sexuales secundarios. HIPOGONADISMO FEMENINO GENERALIDADES Pubertad ETAPA en la cual se alcanza la capacidad reproductora marcada por maduración de organos genitales y desarrollo de caracteres sexuales secundarios, primera menstruación. Ciclo menstrual culminación de fenómenos con la liberación del oocito secundario listo para ser fertilizado, se acompaña de modificaciones de la mama, piel, SNC, cardiovascular, insulina y el hueso. Estrógeno progesterona Fenómeno de Retroalimentación Negativa Fenómeno de retroalimentación Positiva (mitad del ciclo) HIPOGONADISMO FEMENINO GENERALIDADES La clínica varia según momento de la aparición y de la causa. Infancia signos como en el S. Turner, contrariamente edad adulta involución de caracteres sexuales secundarios . Clínicamente las alteraciones hipotalámicas- hipofisiarias y las formas congénitas se caracterizan por infantilismo sexual y las formas adquiridas presentan los síntomas de la causa (tumor) y otros síntomas de déficit hormonal.
- 65. HIPOGONADISMO FEMENINO GENERALIDADES Pubertad Retrasada no inicio de desarrollo mamario a los 13 años o habiéndola iniciado no se desarrolla completamente. La amenorrea primaria es otro aspecto relevante. Incidencia no hay valores de estudio nacionales. España 2.5% de adolescentes de un población normal presenta retraso adquirir caracteres sexuales secundarios a edad superior a la que debían tener. AMENORREA PRIMARIA 1) Carencia de menstruación a los 16 años.(nunca menstruo) 2) No aparición de caracteres sexuales a los 14 años. 3) Puede ser carácter funcional u orgánico. 4) Si existen caracteres sexuales secundarios problema orgánico (obstrucción o desarrollo insuficiente de útero o vagina). 5) Falta de caracteres sexuales secundarios problemas hipotalámicos (GnRH), tumores hipofisiarios, craneofaringiomas, quistes Bolsa de Rathke, limita secreción Lh y FSH. AMENORREA PRIMARIA 6) S. Turner cariotipo 45 XO (disgenesia gonadal).Trastornos de difrenciación sexual. déficit de 21 hidroxilasa. AMENORREA SECUNDARIA 1) Cese de las menstruaciones mas de 6 meses en la mujer que mestrua. Más común luego de pubertad asi: embarazo, menopausia, amenorrea hipotalámica, hiperprolactinemia, hiperandrogenismo (SOP). Evaluación Paciente Amenorreica 1) Es anatómico o funcional, congénito o adquirido. 2) Historia Clínica y examen físico. 3) Medición de FSH y LH para clasificarlas en dos categorías: a) Hipogonadismo Hipogonadotropico: FSH y LH normal o bajo--- falla: Hipotálamo o hipófisis. b) Hipogonadismo Hipergonadotropico: FSH y LH alto--- falla :defecto ovarios o eje hipotalamo- hipofisis (tumor hipofisiario productor de Gn RH .
- 66. CAUSAS CONGENITAS HIPOGONADISMO HIPOGONADOTROPICO a) CONGENITO O IDIOPATICO deficiencia de Gn RH- amenorrea primaria, acompaña anosmia S. Kallmann, ligada a X o autosomico dominante o recesivo. b) HIPERPROLACTINEMIA 1) Niveles altos de prolactina (tumores, hipotiroidismo, medicamentos y embarazo). 2) Inhibe el generador de impulsos de Gn RH hipotalamico, se pierde la secreción pulsátil de LH--efectos fase lutea-- cesan ovulaciones ciclo menstrual corto e irregular . c) AMENORREA HIPOTALAMICA 1) Trastornos en el eje generador GnRH hipotalámico, exceso de ejercicio, stress, disminución ponderal altera secreción pulsatil. 2) Pulsos Gn RH cambian cada 90 minutos fase folicular a media hora ovulación luego cada hora hasta 8 horas lutea. 3) Anorexia nerviosa. 4) Disminución de peso estrés, etc. 2.- HIPOGONADISMO HIPERGONADOTROPICO 1) Insuficiencia Ovárica: menopausia antes de los 40 años, destrucción autoinmunitaria, antecedentes de pubertad normal y menstruación regular prematuramente bochornos, irregularidad menstrual y amenorrea. 2) FSH alta (fase folicular temprana), a veces cariotipos mosaicos de Turner menores de 20 años útil. 3) Asociado a otras enfermedades autoinmunes: insuficiencia suprarrenal, tiroiditis autoinmune, diabetes tipo1, anemia perniciosa, esprue tropical y reumatismos. 4) Tratamiento estrógenos con progesterona.
- 67. Síndrome de Ovarios Poliquísticos Descripción por Stein y Leventhal Perfil Clínico: Oligomenorrea Obesidad Hirsutismo Existencia de: Ovarios Poliquísticos INCIDENCIA DE POLIQUISTOSIS OVÁRICA (Adams et al Br J Med 293: 355,1986) Causas % AMENORREA 32 OLIGOMENORREA 87 ABORTO habitual 80 HIRSUTISMO 87 Sindrome de Ovarios Poliquísticos FISIOPATOLOGÍA Disregulación de pulsos de GnRH Transtornos del eje Gn-IGF 1 Hiperactividad de Obesidad Resistencia insulínica
- 68. AMENORREA PRIMARIA FSH CARIOTIPO N ó (+) (-) DISGENESIA GONADAL OOFORITIS AUTOINMUNE INSUFICIENCIA OVARICA E 2 CAUSA UTERINA N ECOGRAFIA PELVICA FSH NORMAL O BAJO BAJO CAUSA UTERINA ECOGRAFIA PELVICA UTERO ( +) UTERO (-) S. ROKITANSKY RESISTENCIA A ANDROGENOS TABIQUES VAGINALES INFECCIONES: TBC MICOSIS HIPOTALAMO HIPOFISIARIO Deficiencia Gnrh PRL Hipopituitarismo Tumores H - H FSH NORMAL O BAJO + E2 BAJO
- 69. AMENORREA SECUNDARIA B HCG (-) LH Test E + P Test PROGESTERON A (X2) + (Test de Progesterona negativo) Test E + P PRL FSH + N PROLACTINOMA S. ASHERMAN TBC ENDOMETRIAL INSUFICIENCIA OVARICA PREM. N o HIPOPITUITARISMO TUMOR H H DEF. AISLADA Gn (Inicio tardio) LH ( Test de Progesterona +) SOP FUNCIONALES ENDOCRINOPATIAS Trastorno Peso. * S. Cushing. ACOS * Acromegalia. Stress * Hipertirodismo Anorexia Nerviosa Ejercicio Malnutrición. Enfermedad Crónica. Hiperprolactinemia. N ó Hermafroditismo verdadero 2/3 son 46 XX, 1/10 46 XY. Hay existencia de epitelio germinal ovárico y testicular. Puede existir un ovario y un testículo o una gonada con aspecto histológico de ambos. Las 2/3 partes se desarrollan como varones, ginecomastia en el 75%, habitualmente útero y sexo uorgenital Pseudohermafroditismo femenino: HSC y alteraciones en el desarrollo de estructuras mullerianas: ausencia de vagina asociado o no a un útero hipoplásico (síndrome de Mayer- Rokitansk). 46 XX amenorrea primaria
- 70. 1. Mujer de 59 años con cuadro de 2 meses caracterizado por fatiga, malestar general y pérdida de peso, luego se agrega dolor abdominal, vómitos, mareos, y sincopes. Examen: PA 80/50 mmHg, FC 100X'; pálido; presencia de hiperpigmentación. Laboratorio: hiponatremia moderada, hipoglucemia y cortisol 10 /100 ml luego de estimulación con ACTH. ¿Cuál sería el tratamiento a seguir? ERM 2022 a) Levotiroxina b) Dimenhidrinato c) Cloruro sodio 3 % d) Dextrosa 5% e) Corticoides 2. Mujer de 48 años consulta por debilidad, astenia, hiporexia y pérdida de peso desde hace 3 meses. Antecedente de tuberculosis a los 20 años. Examen: PA 80/60 mmHg rítmicos, hipofonéticos. Laboratorio: sodio 130 mEq/L. ¿Cuál es el diagnóstico más probable? ERM 2021 a) Insuficiencia suprarrenal primaria b) Panhipopituitarismo c) Desnutrición severa d) Hipotiroidismo subclínico e) Depresión crónica DISCUSIÓN N 3 DE ENDOCRINO 3. Varón de 29 años, presenta desde hace 3 años hipertensión de inicio súbito que no corresponde bien al tratamiento con 3 fármacos. Al examen: PA: 180/95 mm Hg. Lab: Hipokalemia e hipernatremia. ¿Cuál es el diagnóstico más probable? ENAM 2022 a) Síndrome carcinoide b) Feocromocitoma c) Síndrome de Cushing d) Hiperaldosteronismo primario 4. En el momento del nacimiento se aprecia que un niño presenta genitales externos ambiguos; no existe pene y el clítoris se encuentra significativamente aumentado. La evaluación cromosómica muestra un genotipo XX. Se descubre que tiene ovarios pero no testículos. Las pruebas confirman la ausencia congénita de la enzima corticosuprarrenal -hidroxilasa. ¿Cuál sería el tratamiento adecuado? ERM 2022 a) Corrección quirúrgica y tratamiento hormonal b) No requiere intervención hasta esperar desarrollo hormonal c) Sólo tratamiento hormonal d) Sólo tratamiento quirúrgico e) Conducta expectante 5. Una madre trae a consulta a su hija de 16 años por presentar retraso en la menarquia. Al examen: talla 145 cm, cuello ancho, vello púbico y mama en estadio I de Tanner, pezones mamarios separados. ¿Qué examen solicitaría para confirmar su diagnóstico? ENAM 2022 a) Hormona de crecimiento b) Hormonas tiroideas c) Hormona luteinizante d) Estudio de cariotipo 6. Mujer de 86 años, con hipotiroidismo en tratamiento, es traída a Emergencia por deterioro del estado general progresivo, los familiares refieren que hace tres días, se le administró laxantes salinos por estreñimiento hasta conseguir deposiciones líquidas. Al examen: PA 100/55 mmHg C, rubor facial, reflejos hipoactivos. EKG: prolongación intervalo pR, ensanchamiento qRs. ¿Cuál es el diagnóstico probable? ENAM 2022 a) Hipertiroidismo medicamentoso b) Accidente cerebro vascular c) Encefalopatía hipóxica d) Hipermagnesemia 7. Mujer de 20 años es traída a emergencia por incremento de debilidad de extremidades inferiores que se inició hace 1 semana. Antecedentes: hipertensión arterial diagnosticada hace 2 meses. No recibe tratamiento. Examen: PA: 160/100 mmHg; FC: 90 X'; FR: 20 X'. Despierta, disminución de fuerza muscular en miembros inferiores y reflejos osteotendinosos disminuidos. Laboratorio: pH 7.5, HCO3-: 38 mEq/l, pCO2: 57 mmHg, Na+: 139 mEq/l, K+: 2.3 mEq/l. TAC de abdomen: lesión nodular en glándula adrenal izquierda. ¿Dónde se sintetiza la hormona cuyo aumento explica el cuadro clínico antes descrito? ERM 2020 a) Zona glomerulosa b) Médula adrenal c) Zona reticular d) Cápsula adrenal e) Zona fascicular 8. Varón de 58 años, hace 6 meses presenta epigastralgia y deposiciones diarréicas abundantes. Antecedente de tumor de hipófisis. Endoscopía alta: múltiples úlceras en primera porción de duodeno; además del tumor hipofisario. ¿En qué otros tumores se deben sospechar? ERM 2020 a) Tiroides e hipotálamo b) Páncreas y paratiroides c) Paratiroides y tiroides d) Tiroides y gónadas e) Páncreas y glándulas suprarrenales
- 71. 9. Varón de 50 años con tuberculosis avanzada, desarrolló signos de insuficiencia suprarrenal aguda severa por enfermedad de Addison. ¿Cuál es indicación terapéutica más adecuada? ERM 2020 a) Aldosterona y fludrocortisona b) Metilprednisolona y triamcinolona c) Triamcinolona y dexametasona d) Hidrocortisona y fludrocortisona e) Dexametasona 10. ¿En qué síndrome es más frecuente la enfermedad valvular aórtica bicúspide? ERM 2020 a) Noonan b) Proteus c) Marfan d) Williams e) Turner 11. Una mujer de 18 años de edad de baja estatura, con cuello corto, linfedema de las extremidades inferiores, malformaciones óseas, pecho ancho con los pezones muy separados y ausencia de maduración sexual. Acude preocupada porque no ha menstruado nunca. Se trataría de un síndrome de: ERM 2014 a) Turner. b) Down. c) Klinefelter. d) Angelman. e) Prader - Willi. 12. El síndrome de Kallman 2013 a) Primaria que acompaña al hipogonadismo hipogonadotrófico. b) Secundaria e hipogonadismo hipergonadotrófico. c) Secundaria con ovarios normales y ausencia de GnRH. d) Primaria con concentraciones normales de gonadotropina. e) Secundaria por ausencia de neuronas de GnRH. 13. ¿Cuáles son las características clínicas del síndrome de Cushing? ERM 2012 a) Debilidad muscular distal y estrías en piel. b) Atrofia muscular y palidez. c) Obesidad central o centrípeta y plétora facial. d) Estrías cutáneas violáceas e hipotensión arterial. e) Hirsutismo y palidez generalizada. 14. Mujer de 40 años que presenta astenia, debilidad, náuseas y vómitos desde hace un mes. Al examen: hipotensión y coloración oscura de la piel. El examen de laboratorio muestra hiponatremia. ¿Cuál es el diagnostico más probable? ERM 2012 a) Hipotiroidismo. b) Insuficiencia renal crónica. c) Enfermedad de Addison. d) Cirrosis hepática. e) Insuficiencia cardiaca. 15. ¿Cuál de las siguientes alteraciones NO corresponde a una acromegalia? (ENAM) a) Macroglosia. b) Aumento del volumen de las manos. c) Prognatismo. d) Crecimiento longitudinal de los pies. e) Hipertelorismo. 16. Mujer de 49 años, multípara y obesa, cefalea de 2 meses. Destaca PA 155/90 mmHg. En RM selar una herniación de aracnoides a través del diafragma selar, sin evidencia de tumor. Analítica hormonal normal excepto una reserva de GH patológica y PRL 52 ng/mL. Señale la combinación diagnostico-tratamiento más correcta: a) Adenoma hipofisario no funcionante cirugía transesfenoidal. b) Cefalea tensional tratamiento de la hipertensión arterial. c) Déficit primario de GH tratamiento hormonal sustitutivo. d) Síndrome de silla turca vacía tranquilizar a la paciente. e) Microprolactinoma cabergolina.
- 72. 17. Mujer de 25 años de edad, talla 150 centímetros, peso 80 Kg, con facies pletórica, hirsutismo, debilidad muscular proximal. PA: 160/100 mmHg, glucosa en ayunas 120 mg/dL. El diagnóstico es: (ENAM) a) Diabetes mellitus. b) Obesidad exógena. c) Síndrome de Turner. d) Polimiositis. e) Enfermedad de Cushing. 18. La causa más frecuente de síndrome de Cushing por exceso de ACTH es: a) Secreción de ACTH por tumor de células pequeñas. b) Hiperplasia nodular suprarrenal. c) Microadenoma hipofisario (< 1cm). d) Macroadenoma hipofisario (>1 cm). e) Hiperplasia difusa de células corticotropas. 19. Varón de 28 años de edad presenta episodios de cefalea, diaforesis y palpitaciones desde hace 6 meses. Niega uso de drogas y de historia familiar de HTA. PA 180/150. Resto del examen normal. ¿Cuál es la presunción diagnostica? (ENAM) a) Feocromocitoma. b) Enfermedad de Cushing. c) Crisis tirotóxica. d) Neoplasia endocrina múltiple. e) Hiperplasia de glándula suprarrenal bilateral. 20. El pico máximo de LH, en un ciclo menstrual, se debe a retroalimentación positiva por: ESSALUD a) Progesterona. b) Estrógenos. c) TSH. d) FSH. e) ACTH. 21. ¿Cuál es el orden de mayor a menor de la potencia estrogenica? ESSALUD a) Estriol, estrona, estradiol. b) Estrona, estradiol, estriol. c) Estradiol, estrona, estriol. d) Estradiol, estriol, estrona. e) Estriol, estradiol, estroma. 22. Todas las siguientes son causa de hipogonadismo hipogonadotropo, EXCEPTO: a) Anorexia nerviosa. b) Síndrome de Sheehan. c) Síndrome de Turner. d) Síndrome de Kallman. e) Síndrome de Prader-Willi. 23. Una niña que en la Escala de Turner tiene las siguientes características: mama aumentada de tamaño y areola aumentada sin doble contorno, vello pubiano rizado. ¿Cuál es el estadio? ENAM R a) I. b) IV. c) III. d) V. e) II. 24. Varón de 20 años de edad, que acude por no haber iniciado la pubertad. Tiene testículos blandos y pequeños, micropene, olfato disminuido, testosterona sérica 0,7 ng/mL (N 3-10), LH 2 mUI/mL (N 2-12); PRL 7 ng/mL (N 2-20); RMN: hipófisis normal. El diagnóstico corresponde al síndrome de: (ENAM) a) XO. b) Lawrence Moon Biedl. c) Klinefelter. d) Noonan. e) Kallman. 25. En examen físico a un varón de 17 años se le nota solo desarrollo sexual secundario mínimo, ginecomastia, y un hábito eunocoide alto. Su análisis cromosómico 47 XXY. Síndrome para estos hallazgos: a) De Down b) De Edward c) De Klinefelter d) De Turner e) Multi X