Descargado 381 veces














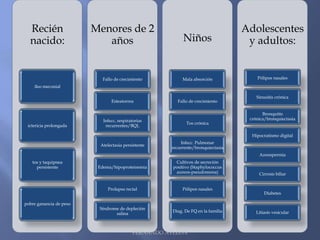
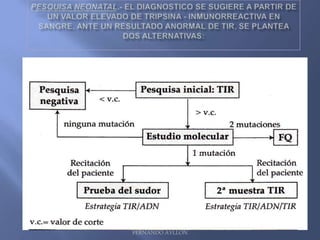


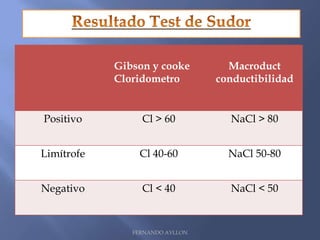

















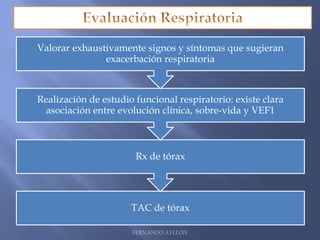








Este documento resume la fibrosis quística, una enfermedad genética hereditaria caracterizada por disfunción de las glándulas exocrinas. Afecta aproximadamente a 1 de cada 6,100 recién nacidos y se debe a mutaciones en el gen CFTR. Los síntomas incluyen enfermedades respiratorias e intestinales, y el diagnóstico se confirma mediante la prueba del sudor. El tratamiento se centra en controlar las infecciones pulmonares y mejorar la nutrición.

























![Fibrosis quistica[1] uuuu sbui](https://cdn.slidesharecdn.com/ss_thumbnails/fibrosisquistica1uuuusbui-100622220522-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)









































