


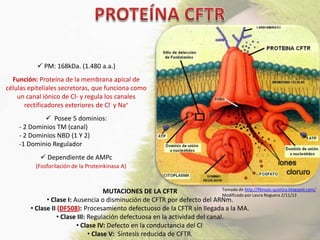
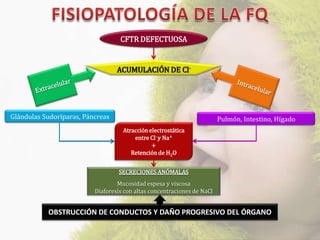


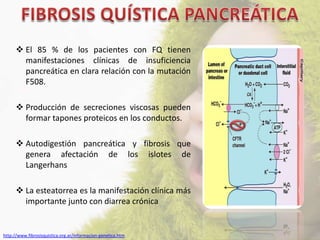


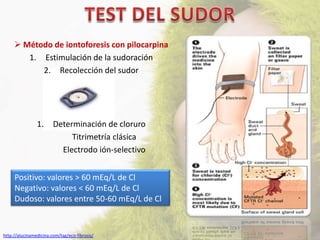



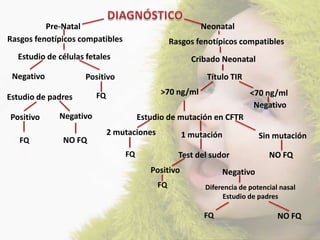


La fibrosis quística (FQ) es una enfermedad genética autosómica recesiva caracterizada por la producción de moco espeso y viscoso, que causa obstrucción en los órganos. La incidencia es de 1/2,500 a 1/3,500, siendo más común en la raza caucásica, y se diagnostica a través de métodos como el test del sudor y análisis genéticos. Las mutaciones en el gen CFTR son responsables de la enfermedad, siendo la más común la DF508, que afecta la función de las células epiteliales, especialmente en los pulmones y el páncreas.























































































