Descargado 653 veces









































![Bibliografía
1.-Programa Nacional de Fibrosis Quística Orientaciones Técnicas Programáticas para
Diagnóstico y Tratamiento 2012.
2.-TraTado de Fibrosis Quística , A. Salcedo Posadas S. Gartner, R.M. Girón Moreno M.D. García
Novo (sociedad española de fibrosis quística.
3.-AVANCES EN FIBROSIS QUÍSTICA UPDATE IN CYSTIC FIBROSIS :DR. ÓSCAR FIELBAUM
C.Departamento de Pediatría, Clínica Las Condes. [REV. MED. CLIN. CONDES - 2011; 22(2) 150-
159]
.](https://image.slidesharecdn.com/fibrosisquistica-160620020635/85/Fibrosis-quistica-47-320.jpg)

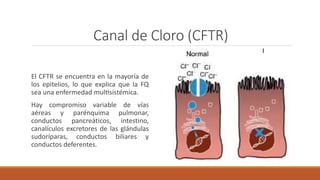
La fibrosis quística es una enfermedad genética hereditaria que afecta principalmente los pulmones y el páncreas. Se hereda de forma autosómica recesiva y se debe a mutaciones en el gen CFTR que codifica un canal de cloro. Esto produce secreciones anormalmente espesas que obstruyen los pulmones y dificultan la digestión. El diagnóstico se realiza mediante el test de sudor o estudios genéticos. El tratamiento incluye kinesiterapia respiratoria, antibióticos y nutrición para mejor































































































