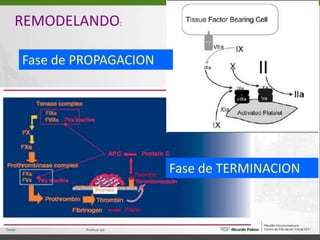
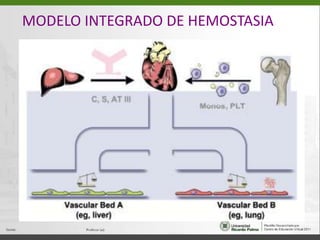
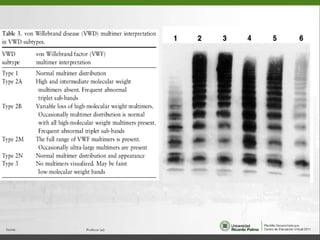
Este documento resume diferentes trastornos de la coagulación hereditarios como la hemofilia A, hemofilia B y la enfermedad de von Willebrand. Describe las características clínicas, el diagnóstico y el tratamiento de cada uno. Explica que la hemofilia A y B se deben a deficiencias del factor VIII y IX respectivamente, mientras que la enfermedad de von Willebrand involucra defectos en la proteína von Willebrand.