Descargado 105 veces





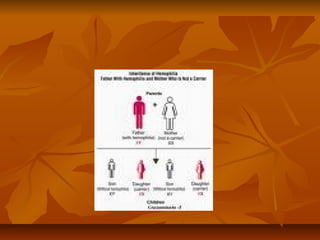













La hemofilia es una enfermedad genética ligada al cromosoma X que causa dificultad en la coagulación de la sangre debido a la deficiencia de factores de coagulación como el factor VIII o IX. Se caracteriza por hemorragias internas y externas. Es hereditaria y se transmite de madres portadoras a hijos. El tratamiento consiste en reemplazar los factores de coagulación deficientes mediante infusiones intravenosas para prevenir y controlar hemorragias.
































































