Descargado 24 veces

















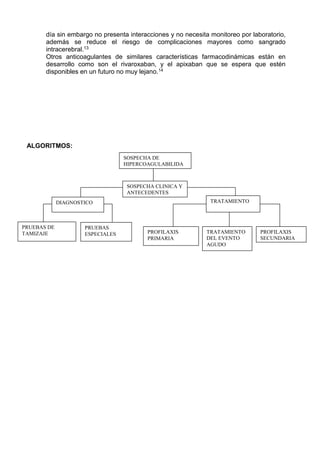
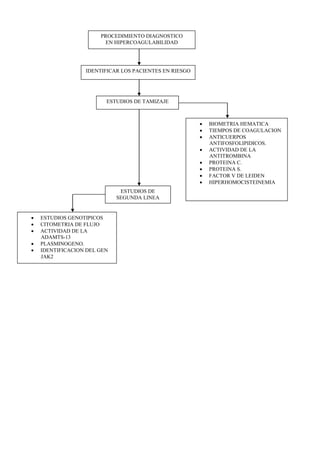
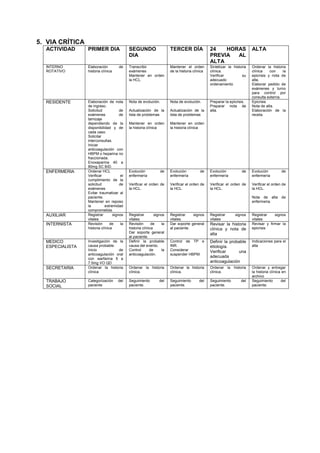


Este documento describe varios trastornos de hipercoagulabilidad, incluyendo la mutación del factor V de Leiden, la mutación 20210 asociada a la protrombina, deficiencias de proteína C y S, deficiencia de antitrombina, síndrome de anticuerpos antifosfolipídicos, niveles elevados de factor VIII, hiperhomocisteinemia, desórdenes mieloproliferativos, y hemoglobinuria paroxística nocturna. Define cada trastorno, su prevalencia, mecanismo y



















































































