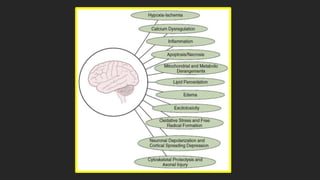
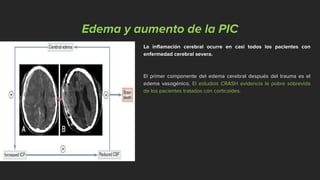
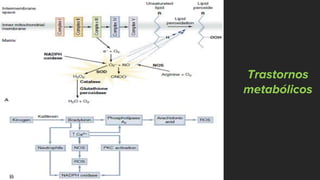
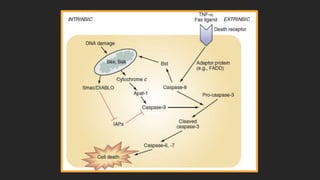
Este documento describe los mecanismos de lesión primaria y secundaria en el trauma craneal. La lesión primaria incluye daño neuronal, axonal y vascular causado por las fuerzas mecánicas del trauma. La lesión secundaria involucra una serie de eventos moleculares y celulares como hipoxia, excitotoxicidad, desregulación del calcio, estrés oxidativo e inflamación que ocurren después de la lesión primaria. Estos procesos secundarios pueden causar daño adicional al tejido cerebral a


















![traumacraneoencefalicoTCE NUEVO [Autoguardado].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/tcenuevoautoguardado-240221213118-eb4712b6-thumbnail.jpg?width=640&height=640&fit=bounds)




















































