Descargado 320 veces






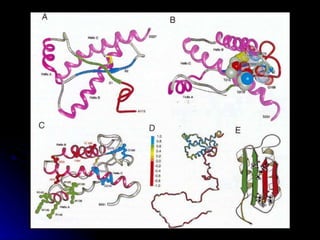































Los priones son partículas acelulares compuestas principalmente por proteínas que causan enfermedades neurodegenerativas llamadas encefalopatías espongiformes transmisibles. Se caracterizan por inducir un cambio en la estructura de proteínas celulares normales que luego se acumulan en el sistema nervioso central causando daño. Aunque no contienen ácidos nucleicos, los priones son transmisibles y pueden adoptar distintas cepas. No existe cura para las enfermedades relacionadas a priones.








































































