Descargado 25 veces














![TRANSPORTE HACIA EL CEREBRO[EDITAR]
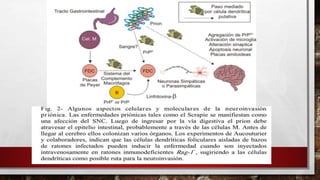
• A PARTIR DE UNA INGESTA CONTAMINADA, EL AGENTE PATÓGENO ES TRANSPORTADO POR
EL EPITELIO INTESTINAL, DESDE DONDE ENTRAA LAS CÉLULAS M (ESPECIALIZADAS EN EL
TRANSPORTE DE MACROMOLÉCULAS Y PARTÍCULAS A TRAVÉS DE LAS CÉLULAS DEL
EPITELIO INTESTINAL) MEDIANTE TRANSCITOSIS. A CONTINUACIÓN, EL AGENTE ENTRA
DENTRO DE LAS CÉLULAS MIGRATORIAS Y DE LOS MACRÓFAGOS (SISTEMA INMUNITARIO).
UNA VEZ RECONOCIDO, SE SINTETIZA UN ANTICUERPO CONTRA EL PRION, PERO NO TIENE
NINGUNA EFICACIA, CAUSA POR LA CUAL EL PRION ES TRANSPORTADO POR EL SISTEMA
INMUNITARIO Y SE ACUMULA EN EL BAZO Y LOS GANGLIOS LINFÁTICOS, QUE ESTÁN MUY
INERVADOS. ESTE HECHO PRODUCE EL CONTAGIO AL TEJIDO NERVIOSO Y LA
CONSECUENTE MUERTE NEURONAL Y POR ELLO, EL CEREBRO ADQUIERE UN ASPECTO
ESPONJOSO](https://image.slidesharecdn.com/prionesyviroides-161003205522/85/Priones-y-viroides-16-320.jpg)









Este documento resume las características de los viroides, priones y sus efectos en plantas y animales. Los viroides son agentes infecciosos similares a los virus pero compuestos solo de ARN sin proteínas o lípidos. Causan enfermedades en plantas. Los priones son proteínas plegadas erróneamente que causan encefalopatías neurodegenerativas en animales al replicarse. Ambos son los agentes infecciosos más simples conocidos y se replican de forma autónoma sin necesidad de material genético.























































































