Este documento presenta la lista de autores del libro "Virología Clínica". Incluye los nombres, títulos y afiliaciones institucionales de más de 50 autores provenientes de universidades e instituciones de Chile, Argentina, Uruguay, México, Bolivia y Estados Unidos. El prólogo describe los objetivos del libro, que son acercar los aspectos básicos y aplicados de la virología para estudiantes y profesionales de la salud, explicando de manera comprensible los virus más relevantes para la salud humana.













![VIROLOG[A ClÍNICA
]~ cf @
*
Vi roides
priones
YCélulas Células Bacteria Poxvirus Virus Proteínas Pequeñas Átomos
vegetales animales
,----,1
ribosomas moléculas
¡----J ¡----J r------1 n
11111 1 1 1 111111 11 1 111111 11 1 111111111 111111 11 1 1111111 11 111111 11 1 1111111 1 1 Metros
10-2
(lcm)
10-3
(lmm)
Microscopio de luz
10-4 10-5 10-.;
(lpm)
Microscopio electrónico
10-7 10-,11 10-9 10-10
(lnm) (lÁ)
Rayos X
Resonancia nuclear
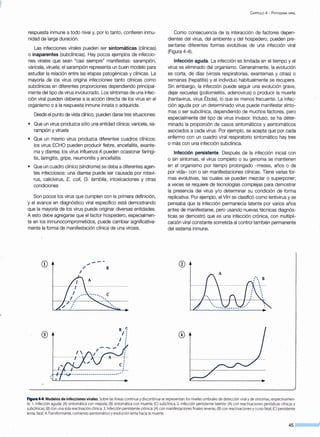
Figura 1-2. Tamaño comparativo de los virus en relación aotros seres vivos, sus componentes estructurales yla capacidad de visualizarlos.
mación genética en forma horizontal. Es fascinante descubrir
cómo un microorganismo tan pequeño y relativamente simple,
se las ingenia para invadir y dominar una célula específica y
multiplicarse para mantener su especie, utilizando las diversas
estrategias que vienen definidas en su pequeño genoma. Y
que además, algunos causen enfermedades, a veces devas-
tadoras. Ante esta realidad resulta ocioso discutir si los virus
son entes vivos.
En resumen, podemos considerar a los virus -forma mínima
y eficiente de vida- como agentes infecciosos relevantes y
como una magnífica herramienta de estudio en biología.](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-14-320.jpg)






































![ViROLOGÍA ClÍNICA
Tabla 7-2. Definiciones operativas en epidemiología
Endemia
Epidemia
Pandemia
Emergencia y
reemergencia
Incidencia
Tasa de ataque
Prevalencia
Mortalidad
Letalidad
Infección presente constantemente, en un nivel significativo. Ej.: herpes simplex, virus papiloma humano, resfrío común,
hepatitis B.
Aumento "inusual" de casos en una comunidad. El concepto de inusual es arbitrario y relativo: tres casos de poliomielitis
o de Ébola pueden significar epidemia, mientras que cien casos de influenza en un país no lo constituyen. Ej.: sarampión,
influenza, dengue, fiebre amarilla.
Epidemia que afecta simultáneamente avarios continentes. Ej.:influenza H1 N1 2009, SARS, VIH.
Aparición de nuevos agentes patógenos o resurgimiento de antiguos aparentemente controlados (Ej.: SIDA, fiebre amarilla,
dengue). Afecciones antiguas cuyo reciente diagnóstico las ha puesto en evidencia, como hantavirus (Capítulo 23: Virus -
emergentes yreemergentes).
Número de casos nuevos detectados en un período (un año, una semana), dividido por la población del lugar determinado
y multiplicado por un amplificador. La incidencia es el resultado de la interacción del agente con la susceptibilidad de la
población, la transmisibilidad de la infección, y de la relación entre infección aparente y subclínica (Figura 7-1 ).
Tasa de incidencia en un período breve que se usa para enfermedades agudas de corta duración (Figura 7-2).
Número de infecciones específicas (clínicas o subclínicas) detectadas en un momento determinado, dividido por la
población del lugar y multiplicado por un amplificador. Se usa para procesos crónicos o estados inmunitarios y para
vigilar algunas infecciones (Ej.:seroprevalencia de anticuerpos anti-sarampión o hepatitis Ben un país, portación de virus
papiloma humano, vigilancia de VIH) (Figura 7-1 ).
Número de muertes de una enfermedad ocurridas en un período, dividido por la población del lugar determinado y
multiplicado por un amplificador. Representa el riesgo de morir por esa condición en esa comunidad. Ej.: la mortalidad por
influenza estacional fue en Chile de 0,89 y 0,21 por 100.000 habitantes en 1999 y 2000, respectivamente.
Número de muertes de una enfermedad ocurridas en un período, dividido por el total de casos de esa enfermedad
ocurridosen ese período; se puede expresar como porcentaje. Es un índice de la gravedad o severidad clínica. Ej.:el
hantavirus tiene una letalidad del 50%, y la influenza AswH1N1del O,1%al 2%.
Tabla 7-3.Los diez pasos de una investigación de un brote epidémico
1 Determinar la existencia de la epidemia
2 Confirmar el diagnóstico
3 Definir el caso y contar los casos
4 Orientar los datos en términos de tiempo, lugar y persona
5 Determinar quién está en riesgo de enfermarse
6 Desarrollar una hipótesis explicando la exposición específica que causó la enfermedad y probarla mediante métodos estadísticos apropiados
7 Comparar la hipótesis con los hechos establecidos
8 Planificar un estudio sistemático
9 Preparar un informe escrito
1O Ejecutar las medidas de prevención y control .
Fuente:Gregg M, New York Oxford University Press, 2002.
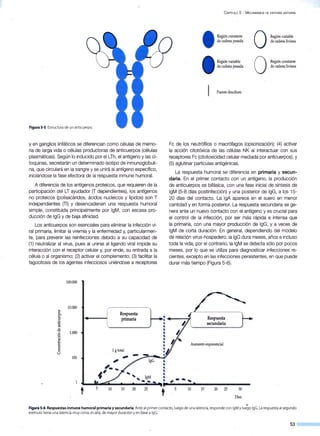
INCIDENCIA YPREVALENCIA
J
Figura 7-1.Presencia de una infección en una población determinada en
un tiempo (incidencia) o momento (prevalencia) delimitados.
68
lOO
90
80
70
60
50
40
30
20
10
o
INCIDENCIA
SUSCEPTIBLES X INFECTADOS X CaSOS clínicos
.,
"-- " -------
't
~(f
J J-=-=-==- -f]-
Tasa Población Susceptibles Infectados Enfermos
de ataque total
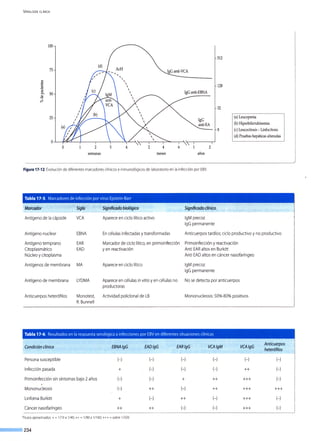
Figura 7-2.La incidencia o tasa de ataque depende de la interacción de factores inheren-
tes al virus(proporción infectados/enfermos) y a la población (susceptibilidad).](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-56-320.jpg)




![l
,~
[
[
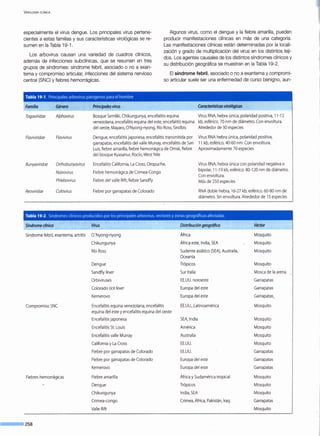
infección nosocomial. El número de contagiados dependerá
de la contagiosidad de la virosis, de la proporción de casos
sintomáticos y subclínicos propia de ella y del número de
susceptibles. El índice de transmisibilidad (Ro) es importan-
te para predecir la evolución -intensidad y duración- de un
brote. El número reproductivo básico (Ro) alude al número de
casos secundarios que un caso generará en el curso de su
enfermedad en una población susceptible. Por ejemplo, se
ha descrito un índice de 18-20 para sarampión, de 1,3 para
influenza estacional y de 1,4 para la gripe A H1 N1 2009 en
México.
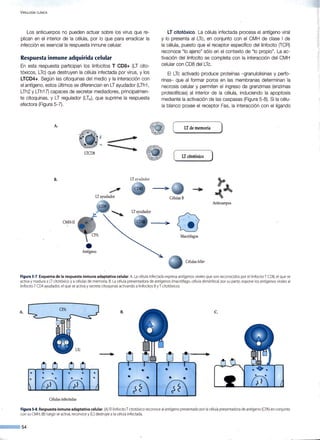
120
~ 100
..§
..o 80<e
...e::
:-;::1
e 60¡:::
Q)
·o
t5 40
0...
ffi
~ 20
Años
A. Incidencia de poliomielitis en Chile:1941-1975
140
120
j 100
~
]
80
¡:::
60·O
t50...
40
ffi
~
20
C APÍTULO 7 - l os VIRUS Y LA COMUNIDAD
Endemias. Corresponden a la presencia permanente del
agente en la comunidad, en cualquier proporción. Por ejem-
plo, la prevalencia de hepatitis B es alta en África y Asia, en
comparación con otros continentes, y siempre es endémica; la
infección por VIH ya es endémica en todo el mundo.
Patrón mixto (endemia-epidemia). Algunas infecciones que
tienen estacionalidad, cada año reaparecen con más o menos
fuerza dependiendo de la inmunidad adquirida y de las varia-
ciones del agente (Ej.: influenza, VRS). Incluso para algunos
virus se han descrito ciclos epidémicos en eras pre y post va-
cunaciones (Ej.: rubéola, sarampión, parotiditis) (Figura 7-3).
Fuente: Anuario enfermedades de denuncia obligatoria. Ministerio de Salud, Chile.
0+-~--.-----.-~--.--,--,--,--,--,--,--,--,--,--,--,---.
~~~~~~~~~~~~~~~~~~~
B. Incidencia de rubéola en Chile: 1980-1998
250
Vacuna
</)
200
§
~
150...e::
]
-~ 100
t50...
ffi 50~
1970 1972 1974 1976 1978 1880 1882 1884 1886 1888 1990 1992 1994 1996 1998 2000 2002 2004 2006
Años
C. Incidencia de parotiditis en Chile: 1970-2006
Figura 7-3. Efecto en la incidenciaen Chile de lavacunade tres infecciones virales: poliomielitis (A), rubéola(B) y parotiditis(C).
73](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-61-320.jpg)















![CAPÍTULO 9
Control de infecciones virales
Luis Fidel Avendaño
La necesidad del hombre de sobrevivir como especie le ha
compelido a diseñar estrategias para controlar los agentes
que pudieran afectarlo, tales como los virus. El avance de la
civilización ha traído cambios en el ecosistema a los que tan-
to los virus como sus hospederos no han estado ajenos. Los
progresos de la ciencia y la tecnología en diversos aspectos
-urbanización, transportes, costumbres, educación, comu-
nicaciones, etc.- han permitido desarrollar estrategias para
convivir con microorganismos potencialmente patógenos cuyo
contacto parece inevitable.
Pueden mencionarse siete grandes estrategias destinadas
a controlar los agentes infecciosos: educación, modificación
del medio ambiente, bioseguridad, esterilización y desinfec-
ción, antivirales, quimioprofilaxis y vacunas. En este capítulo se
hará referencia sucintamente a algunas de ellas, sin incluir los
antivirales y las vacunas, puesto que se tratan en profundidad
en otros capítulos; igualmente, los aspectos de control espe-
cífico de las infecciones se abordarán en los capítulos corres-
pondientes a los virus que requieran menciones especiales.
EDUCACIÓN
La educación es indispensable en cada una de las estrate-
gias de control de infecciones, pero su eficacia depende de
muchos factores, como la transmisibilidad del agente, de los
mecanismos de contagio y de la patogenia de la infección,
entre otros. Debe estar siempre asociada a otras estrategias
de control para lograr las metas, pues implica no sólo la ad-
quisición de información, sino también la interiorización de la
misma reflejada en cambios de hábitos y actitudes, que son en
última instancia los que permiten concretar las acciones y los
procedimientos. Por eso, su efectividad en el control de las in-
fecciones varía si sus contenidos no se mantienen y refuerzan
en el tiempo.
Por ejemplo, la educación sobre la necesidad del lavarse las
manos con agua y jabón, el uso de zonas limpias y sucias para
Contenido
Educaci9n _____________________________________]_Q
Medio ambiente 90
___§~~~~9-~!!~~----------------------------------------------------------------------------------------------------------~~-
-~~_r:!~~~_ción ~~sinfecci~~-antisee~J-~ _____9~
---~~f!2i_~ec~!!!~~~-------------------------------------------------------------------------------------~-~
la preparación de los alimentos, la adecuada cocción de los
mismos, más medidas de saneamiento ambiental (acceso a
agua potable y alcantarillado) son intervenciones con un fuerte
contenido educativo y efectivas para el control de las infeccio-
nes de transmisión fecal-oral.
Las recomendaciones sobre el manejo de las secreciones
en las virosis transmitidas por la vía respiratoria son menos
efectivas, pues aunque un caso sintomático "se cubra la boca
al toser", los ambientes cerrados son propicios para la trans-
misión viral por las secreciones expelidas por casos asintomá-
ticos al hablar o simplemente respirar.
El control de las infecciones transmitidas por vía sexual re-
presenta un desafío por la existencia de infecciones crónicas
asintomáticas como fuentes de contagio y por la importancia
que la sociedad le otorga al acto sexual.
Incluso si se acepta que las vacunas son los medios más
efectivos para controlar muchos agentes, su aplicación tam-
bién requiere de educación, para que el público acepte y pro-
mueva su uso y las autoridades comprendan sus ventajas y
financien su implementación, como ocurrió con la pandemia
de influenza de 2009.
MEDIO AMBIENTE
Como la población humana ha ido creciendo, necesariamente
ha habitado nuevos territorios, alterando el carácter "silvestre"
de la naturaleza. Desde que el hombre cambió su condición
de nómade a sedentario (1 0.000 años a.C.), cultivó vegeta-
les y domesticó animales, cambió su relación con los agentes
microbianos naturales. En nuestro caso, aumentaron las ins-
tancias de intercambio y la influencia sobre virus de plantas y
de animales silvestres, aparte de los virus propios de los seres
humanos. En la medida en que ha aumentado el conocimiento
sobre los diversos virus, el hombre está tratando de controlar el
riesgo de infección y tomando medidas para minimizar las con-
secuencias negativas que ello pudiera traer para la naturaleza.](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-78-320.jpg)












![Antivirales
Luis Fidel Avendaño
E1desarrollo de la terapia antiviral ha debido superar dos
obstáculos inherentes a la naturaleza de la interacción del
virus con el hospedero. Primero, los virus son parásitos intra-
celulares obligados, por lo que los antivirales deben alcanzar
concentraciones adecuadas a nivel intracelular, lo que obliga
en algunos casos a suministrar dosis altas, cercanas a los nive-
les tóxicos. Segundo, los virus carecen de capacidad ener-
gética metabólica propia y utilizan la de la célula, por lo que
se corre el riesgo de que los antivirales actúen también sobre
las células normales. Entonces, su eficacia depende en gran
medida de la selectividad del antiviral, es decir, de la capaci-
dad de discriminar entre procesos virales y celulares.
Los avances en el conocimiento de la estructura viral, de
la genética y de los procesos moleculares involucrados en la
infección viral han permitido definir algunas etapas por las que
pasan las enzimas virales o celulares codificadas por los virus,
que podrían ser inhibidos selectivamente, sin afectar los proce-
sos metabólicos normales.
En general el espectro de acción de los antivirales es res-
tringido. Su uso requiere de un diagnóstico específico y rápido,
condición que se cumple sólo en algunos cuadros clínicos ca-
racterísticos (varicela, herpes zóster, sarampión, etc.) o situacio-
nes epidemiológicas (influenza). Hoy se acepta que la mayoría
de loq cuadros infecciosos virales representan "síndromes" de
etiología múltiple (infecciones respiratorias, diarreas, exante-
mas, etc.) o infecciones persistentes (herpes, citomegalovirus,
hepatitis B y C, VIH, y otros), donde la manifestación de los
síntomas es tardía. Además, la creciente población de indivi-
duos inmunosuprimidos hace más difícil establecer la etiología
CAPÍTULO 11
Contenido
Et~pas_J~ la multipJ~~_ón ~~~~------ ------------------------------------------1Q~
Mecanismos de acción de los antivirales 105
Adsorción: anticuerpos naturales, palivizumab 105
Penetración y denudamiento: amantadina- rimantadina, pleconaril,
inhibidores de la fusión 106
Síntesis de macromoléculas 106
ADN: aciclovir y ganciclovir 106
ARN Antirretrovirales análogos de nucleósidos: zidovudina y otros 109
Antirretrovirales nucleotídicos: tenofovir 11 O
Antirretrovirales no análogos de nucleósidos 11 O
Antirretrovirales inhibidores de la integrasa 11 O
Ribavirina 11 O
Análogos de pirofosfato 11 O
Proteínas: interferón e inhibidores de proteasas 11 O
Liberación viral 111
------~~~_e-~~~-~--~~-~0_~-----------------------------------------------------------------------!2]_
en base a características clínicas. Sin embargo, el avance bio-
tecnológico ha puesto a disposición una variedad de técnicas
para el diagnóstico viral específico -que entregan resultados en
plazo de horas-, lo que facilita el uso racional de antivirales.
La evaluación de la terapia antiviral también es compleja. La
especificidad de la relación virus-célula hospedera dificulta la
extrapolación de resultados obtenidos ín vítro o en animales
a la infección en seres humanos; además, no se dispone de
modelos animales para estudiar algunos virus que sólo afectan
al hombre.
El desarrollo de un antiviral de uso clínico no demora menos
de diez años. En efecto, una vez demostrada la eficacia e ino-
cuidad ín vítro y luego ín vivo en modelos animales, se deben
analizar las características farmacodinámicas y la toxicidad en
humanos, en un número limitado de individuos sanos (fase 1);
posteriormente, se diseñan estudios clínicos controlados en
mayor escala, analizando múltiples parámetros, como metas
de tratar;niento, tamaño de la muestra, esquemas de tratamien-
to, uso de sistemas aleatorios y doble ciego, etc. (fases 2-4).
En la evaluación de la terapia antiviral puede influir la virulencia
del virus, a veces dependiente del tipo, subtipo o genotipo a
que pertenezca la cepa (Ej.: influenza, adenovirus, hepatits C,
VIH, etc.). La respuesta natural del individuo ante la infección, y
por ende al tratamiento antiviral, también depende de su esta-
do inmur:Jitario, que puede ser difícil de medir cuantitativamen-
te. Finalmente, la precocidad del tratamiento es un factor de
éxito fundamental que se logra raramente, pues muchas virosis
se mañifiestan cuando la infección está bastante desarrollada.
Además, algunos grupos de virus -herpes, hepatitis B y C, VIH
103](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-91-320.jpg)


































![damente el 15% de las consultas ambulatorias por diarrea, el
30% de las consultas de urgencia y el 40% de las hospitaliza-
ciones. Asimismo, son los agentes más importantes de diarrea
infantil nosocomial. La incidencia de diarrea por rotavirus en
niños menores de 18 meses de edad es de 0,3 a 0,8 episodios/
niño al año, lo cual indica que la gran mayoría de los niños, al
llegar a los tres años de edad, ya se han expuesto a este virus.
Se ha estimado que cada año mueren en eJ mundo cerca de
600.000 niños por esta infección. Las muertes se concentran
en los países pobres, en donde los niños infectados no reciben
las terapias orales o intravenosas necesarias para prevenir o
tratar la deshidratación.
En la mayoría de los países con clima templado el rotavirus
se concentra en los meses más fríos y prácticamente desapa-
rece en el verano. Esta estacionalidad no es universal, ya que
en Chile y en países con clima tropical como Brasil, el virus
circula todo el año, sin una predisposición estacional definida.
No existen explicaciones claras para este comportamiento epi-
demiológico.
La caracterización antigénica del virus, dirigida especial-
mente a la identificación de las proteínas que podrían inducir
inmunidad protectora, ha sido extensamente estudiada a tra-
vés de técnicas de neutralización viral y mediante el uso de
anticuerpos monoclonales dirigidos contra epítopes especí-
ficos. También se han desarrollado cientos de estudios para
identificar los componentes del sistema inmune humano que
participan en la respuesta a infección y que podrían proteger
contra reinfecciones. El estado actual del conocimiento rela-
cionado con el rotavirus y la inmunidad se puede resumir en
cinco aspectos fundamentales.
Las proteínas VP7 y VP4 están codificadas por genes
distintos. Para VP7 se han descrito catorce tipos G, de los
cuales a la fecha diez infectan al ser humano. Para VP4 se su-
giere la existencia de por lo menos veinte tipos P, de los cuales
se han identificado nueve en el ser humano. De los más de
veinte tipos antigénicos de rotavirus que se han detectado a lo
largo de los años, sólo cinco patrones antigénicos representan
la mayor parte de los virus circulantes en el mundo (tipos P[8]
G1, P[8]G3, P[8]G4, P[8]G9 y P[4]G2), que representan com-
binaciones de cinco tipos VP7 (tipos G) y dos tipos VP4 (tipos
P). La circulación de estos diferentes tipos antigénicos varía de
una región a otra y dentro de una misma zona en diferentes
períodos.
La infección natural confiere protección contra reinfec-
ciones. Si bien un ser humano puede infectarse repetida,men ~
te durante la vida, es la primera infección la que con mayor
frecuencia se asocia a síntomas moderados o severos; las
reinfecciones son en su mayoría leves o asintomáticas, inde-
pendientemente del serotipo viral involucrado.
La infección natural produce un aumento de los anti-
cuerpos séricos del tipo lgM, lgG, e lgA y los anticuerpos
de mucosa tipo lgA. Mayores niveles de estos anticuerpos en
sangre y/o deposiciones se correlacionan con protección con-
tra la infección. "Correlación" no significa necesariamente que
son los responsables directos o únicos de protección.
VP7 y VP4 son los antígenos neutralizantes, es decir,
inducen anticuerpos capaces de neutralizar la infección viral.
La primoinfección induce anticuerpos séricos neutralizantes
C APÍTULO 13 - VIRUS Y DIARREAS
contra el tipo VP7 y VP4 del virus infectante y en menor me-
dida contra VP7 y VP4 diferentes. Las infecciones repetidas
"amplían" esta respuesta a otros serotipos. Esta mayor o me-
nor especificidad de la respuesta inmune contra VP7 y VP4 y
su función en la protección fue el centro dy la discusión para
decidir las estrategias de desarrollo de vacunas en los años
noventa.
La lactancia materna protege contra infección sintomá-
tica por rotavirus probablemente a través de lgA y otros me-
canismos aún no claramente establecidos.
Aún quedan dudas por resolver respecto de la respuesta
inmune. Si bien la inducción de anticuerpos contra VPT y VP4
es un mecanismo fundamental de inmunidad protectora, es
probable que existan otras proteínas/epítopes relevantes que
induzcan protección no necesariamente neutralizante en VP6
o en proteínas no estructurales. Cabría esperar que si NSP4
resulta ser una "enterotoxina", induzca anticuerpos "antitoxi-
na", que podrían ser protectores al momento de·una reinfec-
ción. Tampoco está clara la función de la inmunidad celular
e intestinal en la protección contra las reinfecciones, lo que
actualmente es muy difícil de evaluar. Es probable que ambos
sistemas jueguen un papel importante en el desarrollo de in-
munidad protectora.
La interacción virus-hospedero es compleja: y el resultado
final -ausencia de infección, infección asintomática o sinto-
mática leve, moderada o severa- dependerá de la combina-
ción de interacciones entre inmunidad innata y adquirida, tanto
humoral como celular, así como de factores virales como dosis
infectante y posiblemente dé los tipos antigénicos. Es posible
que la complejidad se deba a que las proteínas virales VP7 y
VP4 interactúan con diversos componentes de la membrana
citoplasmática en forma secuencial. VP4 lo hace inicialmente
con residuos de ácido siálico y posteriormente durante la pe-
netración viral con a1 ~2, para que a continuación VP7 lo haga
con otro grupo de integrinas. Esto explica la dificultfil.d para
determinar el tropismo por diferentes células.
CALICIVIRUS HUMANOS: NOROVIRUS Y
SAPOVIRUS
Luego del descubrimiento del virus Norwalk en 1972, en dife-
rentes localidades del mundo se reportó una amplia variedad
de virus de tamaño y de aspecto similar al microscopio elec-
trónico, relacionados con gastroenteritis aguda, a los que en
muchos casos se dio el nombre de la localidad donde fueron
descubiertos. Entre ellos destaca el sapovirus (denominado
anteriormente virus Sapporo), que fue detectado en 1982 en
un brote en niños que residían en Sapporo, Japón. Los dos
géneros, norovirus y sapovirus, se agrupan dentro de los cali-
civirus humanos (HuCVs), para diferenciarlos de los calicivirus
que infectan exclusivamente a animales.
Los HuCVs son un grupo de virus de ARN de hebra simple
de 7,5 kb de polaridad positiva, icosaédricos, desnudos, de 27
nm de diámetro, morfológicamente indistinguibles pero que se
diferencian genética y antigénicamente entre sí. El genoma del
norovirus presenta tres marcos de lectura abierta (ORF); ORF1
codifica una proteína no estructural, la ARN polimerasa-ARN
dependiente, ORF2 y ORF3 codifican la proteína de cápside
141](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-127-320.jpg)






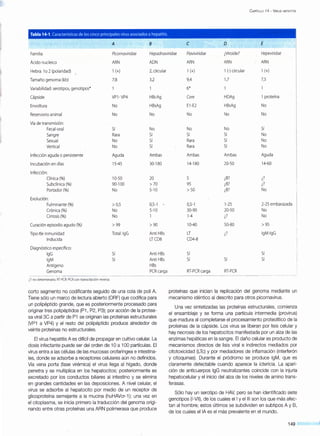




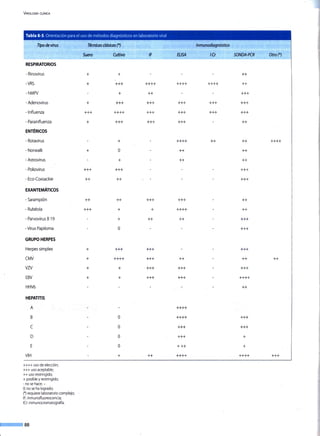
![Figura 14·6. Replicación
del virus hepatitisB.
endocitosis
ADNccc
transcripción
~------------~AAÁ-------'AAÁ
----------'AAÁ
_NV
(Th) y citotóxicos, a diferencia de lo que ocurre en aquellos
pacientes con infección crónica. Esto último explica la alta fre-
cuencia de infección crónica en los neonatos (90%).
Epidemiología
A nivel mundial, se estima que dos mil millones de personas
han sido infectadas por el HBV y que más de 360 millones pre-
sentan una infección crónica y están en riesgo de desarrollar
una enfermedad hepática grave, como cirrosis y hepatocarci-
noma (CHC). Alrededor de un tercio de los casos de cirrosis y
la mitad de los CHC en el mundo, son atribuibles a una infec-
ción crónica por HBV.
El CHC es el quinto cáncer más frecuente y la tercera causa
de mortalidad por cáncer en el mundo. Según estimaciones
de la Organización Mundial de la Salud, se producen 5 millo-
nes de casos de hepatitis aguda anualmente, lo que implica
que el 6% al 10% de ellos desarrollará un curso crónico si la
infección ocurre en la edad adulta, y el 90% si afecta al recién
nacido.
El HBV es un virus de distribución universal. Sin embargo,
la prevalencia de esta infección puede oscilar entre el O,1% y
el 20% en diferentes regiones del mundo. Este amplio rango
de prevalencia de HBV se relaciona directamente con la edad
de la primoinfección, que está inversamente relacionada con el
riesgo de evolución a la cronicidad.
Según datos de prevalencia de HBsAg en dadores de san-
gre de las distintas poblaciones estudiadas, se ha determinado
que hay regiones de endemia alta(~ 8%), intermedia (2% a <
8%) y baja(< 2%). Las áreas de mayor endemia se encuentran
ARNm
Citoplasma
proteínas envoltura
cadena L(-)
CAPÍTULO 14 - V IRUS HEPATITIS
secreción
i síntesis
cadena S(+)
~
l
síntesis
cadena S(+)
o
l
transcripción
reversa
]_----~~- Q
ARN pregenómico
encapsidación
en regiones del sudeste de Asia, África subsahariana, China e
islas del Pacífico. Dentro de las zonas de endemia intermedia
se describen algunos países del sudeste de Europa, cuenca
del Mediterráneo, Oriente medio y parte de Sudamérica. Chile
presenta una endemia baja (0,3%), al igual que algunos países
de Europa occidental, Australia, Canadá, los EE.UU. y Nueva
Zelanda (Figura 14-7).
Las personas con infección crónica por el HBV man-
tienen la cadena de transmisión de este virus. Por lo tanto,
a mayor endemicidad en una región, mayor será el riesgo de
infección de una persona a lo largo de su vida, que puede
llegar hasta el 70%.
La fuente de transmisión fundamental es la sangre contami-
nada, lo que en la práctica se traduce en que todas aquellas
personas con HBsAg positivo son una fuente de contagio po-
tencial. El virus puede detectarse en otros fluidos corporales,
tales como las secreciones vaginales, el semen y la leche ma-
terna principalmente, y en menor grado en el sudor, lágrimas,
orina y saliva. Por lo tanto, cualquier fluido corporal contamina-
do con sangre, puede ser una potencial fuente de transmisión,
especialmente cuando la carga viral es alta.
Las vías de transmisión son básicamente la sexual, verti-
cal/perinatal y parenteral, y la frecuencia de cada una de ellas
depende de la endemicidad de cada región. La principal vía
de transmisión -responsable del 75% de los infectados en
el mundo- en los países con alta endemia es la infección
transplacentaria (transmisión vertical) y/o perinatal desde
una mujer embarazada hacia el recién nacido. Esta infección
se produce en el 90% de los casos cuando la madre tiene una
155](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-140-320.jpg)


























































![Los virus de la familia Herpesviridae representan una forma
particular de regulación de la expresión génica. Existen tres
pulsos de transcripción que promueven la síntesis secuencial
de mensajeros, clasificados en alfa, beta y gama. Los genes
alfa (a) son "inmediatos tempranos" y conducen a la síntesis de
proteínas involucradas en la regulación de la expresión génica
(transactivadores) tanto del virus como de la célula. Los genes
beta (~) "tempranos" contribuyen a la producción de las proteí-
nas principalmente involucradas en la replicación del genoma
viral. Finalmente, los genes gama (y) "tardíos", que representan
aproximadamente el 50%·del genoma, codifican proteínas es-
tructurales de la cápsula y glicoproteínas de envoltura (Figura
17-3), lo que permite el paso a la etapa de ensamblaje, que
transcurre en el núcleo, y mediante la cual las proteínas de la
cápsula se empaquetan alrededor del ADN y se le agregan
proteínas del tegumento.
Las glicoproteínas virales presentes en la membrana nuclear
estimulan la yemación viral a través de la membrana nuclear. Al
parecer, habría un doble proceso de envoltura, pues la nucleo-
cápsula con manto sale del núcleo y se trasloca al citoplasma.
Allí se libera de su ·cubierta, se rodea de más proteínas del te-
gumento y adquiere su membrana definitiva a partir del aparato
de Golgi, lugar donde ya se han insertado más glicoproteínas
de superficie. Completada su maduración, la salida desde la
célula ocurre por exocitosis (yemación retrógrada) luego de la
fusión de la membrana de la vesícula del Golgi con la citoplas-
mática, o bien, por lisis por la muerte celular (Figura 17-4).
En el ciclo de latencia el virus persiste en el núcleo co-
mo un episoma (no integrado) en el núcleo de la célula; los
episomas son ADN circulares que se replican independiente-
mente del ciclo replicativo celular. En esta fase de latencia se
expresan sólo algunos genes, llamados transcritos asociados
a latencia (LAT), cuya función podría ser prevenir la expresión
de otros genes virales que promuevan apoptosis; no hay repli-
cación viral.
Las subfamilias de herpesvirus tienen distintos sitios de la-
tencia. Los virus Alfaherpesvirinae (HSV-1 y -2, VZV) lo hacen
en las neuronas de ganglios sensitivos, mientras que los Beta-
herpesvirinae (CMV, HHV-6 y -7) en células linfoides, riñones,
glándulas salivares y endotelio vascular, y los virus Gama-
herpesvirinae (EBV, HHV-8) establecen latencia en linfocitos
To B.
C APÍTULO 17 - VIRUS HERPES
PATOGENIA
La patología que producen en el ser humano corresponde al
modelo de infección persistente latente. La mayoría se presen-
ta como infecciones subclínicas que se adquieren en la niñez o
juventud; sin embargo, como los virus quedan latentes en dis-
tintos sitios, pueden reactivarse en forma sintomática o sub-
clínica, constituyendo una constante fuente de contagio. Las
manifestaciones de enfermedad dependen fundamentalmente
del herpesvirus involucrado y del individuo, pero en algunos
casos el factor ambiente también juega un rol trascendente.
El creciente número de individuos inmunocomprometidos
(trasplantados, personas con cáncer sometidas a quimiotera-
pia, portadores de VIH/SIDA, etc.) representa una población
en riesgo de reinfección o que los virus latentes se reactiven
y se produzcan enfermedades graves tales como encefalitis,
neumonitis intersticial, rechazo de órganos trasplantados, et-
cétera.
La variedad de síndromes asociados a herpesvirus es am-
plia, por lo que en el presente libro se han presentado en varios
capítulos. Así, el virus herpes simplex 1 puede producir lesio-
nes mucocutáneas de curso benigno, pero también encefalitis
y sepsis neonatal, que implican alta letalidad. Por su parte, una
simple varicela puede complicarse en ·individuos con alteración
del sistema inmune. El EBV, que habitualmente causa la mo-
nonucleosis infecciosa, puede asociarse a tumores malignos.
El CMV pasa habitualmente desapercibido en la infancia, pero
puede provocar cuadros febriles prolongados y diversa pato-
logía en inmunocomprometidos; además, es la principal causa
de infección intrauterina y perinatal.
DIAGNÓSTICO VIRAL
Los alfaherpesvirus crecen rápido en cultivos celulares, su ciclo
de replicación es corto y son líticos en fibroblastos y células
epiteliales. En cambio, la infección de los betaherpesvirus es
más lenta y el ciclo replicativo más largo. Por su parte, los
gamaherpesvirus crecen sólo en células linfoblastoides. En
consecuencia, el diagnóstico de virus herpes simplex es sen-
cillo y rápido en cultivo celular, lo que no ocurre con los otros
herpesvirus. Algunas técnicas de inmunodiagnóstico permiten
identificar antígenos específicos de cada herpesvirus, lo que
facilita la determinación de inmunoglobulinas, permitiendo tan-
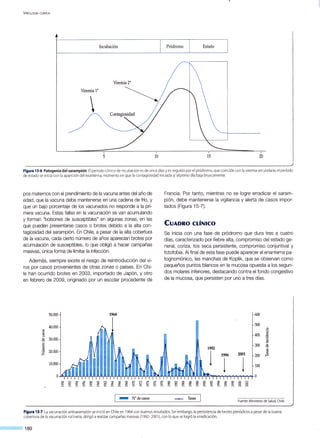
r-·-----------------------------
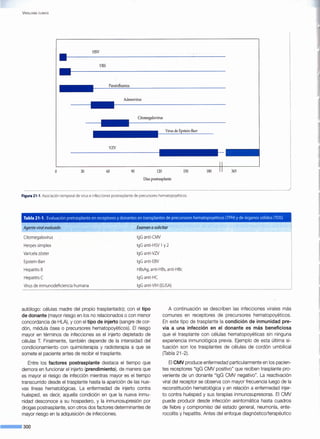
[____c_e_n_es_a___~)• • i Prote::p::atas .,____.,...(, --:- -G-en: ¡-- - ]
/ ' /
Proteínas tardías F----...._: Morfogénesis ymaduración viral
en el núcleo
Figura 17-3.Ciclo replicativo del CMV: expresión de genes y sus fu nciones en la infección productiva.
215](https://image.slidesharecdn.com/virologaclnica-160308120209/85/Virologia-Clinica-200-320.jpg)