El documento describe el metabolismo del hemo y la bilirrubina. El hemo se forma a partir de la protoporfirina III y se une a proteínas como la hemoglobina. La bilirrubina se produce a partir del catabolismo del grupo hemo y se transporta unida a albúmina al hígado, donde se conjuga y se excreta en la bilis. Niveles elevados de bilirrubina causan ictericia.
1. METABOLISMO DEL HEMO
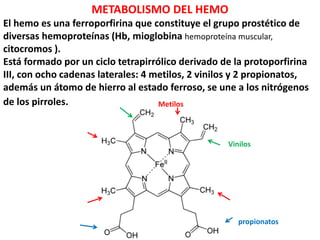
El hemo es una ferroporfirina que constituye el grupo prostético de
diversas hemoproteínas (Hb, mioglobina hemoproteína muscular,
citocromos ).
Está formado por un ciclo tetrapirrólico derivado de la protoporfirina
III, con ocho cadenas laterales: 4 metilos, 2 vinilos y 2 propionatos,
además un átomo de hierro al estado ferroso, se une a los nitrógenos
de los pirroles.
propionatos
Metilos
Vinilos
3. • Hemoglobina A o HbA: llamada hemoglobina del adulto o hemoglobina normal,
representa aproximadamente el 97% de la hemoglobina degradada en el adulto,
formada por dos globinas alfa y dos globinas beta.
• Hemoglobina A2: Representa menos del 2,5% de la hemoglobina después del
nacimiento, formada por dos globinas alfa y dos globinas delta, que aumenta de
forma importante en la beta-talasemia, al no poder sintetizar globinas beta.
• Hemoglobina S: Hemoglobina alterada genéticamente presente en la Anemia de
Células Falciformes. Afecta predominantemente a la población afroamericana y
amerindia.
•Hemoglobina F: Hemoglobina característica del feto.
•Oxihemoglobina: Representa la hemoglobina que se encuentra unida al oxígeno
normalmente ( Hb+O2)
•Metahemoglobina: Hemoglobina con grupo hemo con hierro en estado férrico, Fe
(III) (oxidado). Este tipo de hemoglobina no se une al oxígeno. Se produce por una
enfermedad congénita en la cual hay deficiencia de metahemoglobina reductasa,
la cual mantiene el hierro como Fe(II). La metahemoglobina también se puede
producir por intoxicación de nitritos, porque son agentes metahemoglobinizantes.
4. •Carbaminohemoglobina: Se refiere a la hemoglobina unida al CO2 después del
intercambio gaseoso entre los glóbulos rojos y los tejidos (Hb+CO2).
•Carboxihemoglobina: Hemoglobina resultante de la unión con el CO. Es letal en
grandes concentraciones (40%). El CO presenta una afinidad 200 veces mayor que el
Oxígeno por la Hb desplazándolo a este fácilmente produciendo hipoxia tisular, pero
con una coloración cutánea normal (produce coloración sanguínea fuertemente roja)
(Hb+CO).
•Hemoglobina glucosilada: Aunque se encuentra normalmente presente en sangre
en bajos niveles, en patologías como la diabetes se ve aumentada. Resulta de la
unión de la Hb con carbohidratos libres unidos a cadenas carbonadas con funciones
ácidas en el carbono 3 y 4.
5.
6. Los niveles de Hb por debajo de lo normal pueden deberse a:
• Anemia (diversos tipos).
• Sangrado.
• Deficiencia de eritropoyetina (por enfermedad renal).
• Intoxicación con Pb
• Desnutrición.
• Deficiencias nutricionales de Fe, folato, vit B12 y vit B6
• Sobrehidratación.
• Destrucción de los GR rojos asociada con una reacción a
transfusión.
Los niveles de Hb por encima de lo normal pueden deberse a:
• Enfermedad cardíaca congénita.
• pulmonar
• Aumento en la formación de glóbulos rojos debido a demasiada
eritropoyetina. (médula ósea (síndrome mieloproliferativo), o por una reacción a
bajos niveles de oxígeno crónicos
• Fibrosis pulmonar.
7. BIOSÍNTESIS
El precursor Protoporfirina III se sintetiza a partir de glicina y
succinil CoA
Aa libres
acetoglutarato
d aminolevulinato sintasa
CoA-SH
MITOCONDRIA
CITOPLASMA
Deficiencia enzimática
Anemia sideroblástica hereditaria
HEMO inhibe la enzima
Intoxicación con
Pb inhibe la enzima
8.
9. TRES ETAPAS
1- Síntesis del ácido d aminolevulínico
acetoglutarato Aa libres ALA
d aminolevulinato sintasa
Succinil-CoA + Glicina ácido d aminolevulínico
fosfato de piridoxal - Mg
Principal punto de control
•Hemo, acción alostérica inhibitoria
•Hemoglobina, hemoproteinas correpresores disminuyendo la actividad
vida media de la enzima corta (1hora en hígado)
•Hipoxia, eritropoyetina, hormonas esteroideas, fármacos (barbituricos)
alcohol inducen a la síntesis de la enzima
CoA-SH
10. 2- FORMACIÓN DE PORFOBILINÓGENO
El d aminolevulínico formado en mitocondria pasa al citoplasma
aminolevulinato deshidratasa
2 d aminolevulínico Porfobilinógeno + 2 H2O
ALA deshidratasa ( Zn+2 y grupos sulfhidrilos)
Pb
11.
12. 3- SÍNTESIS DE PORFIRINA:
Polimerizan cuatro moléculas de porfobilinógeno, forman un anillo tetrapirrólico de
uroporfirinógeno. Actúan 2 enzimas: uroporfirinógeno I sintasa y uroporfirinógeno
III cosintasa. Se producen modificaciones de las cadenas laterales
(descarboxilaciones y oxidaciones) y se forma el anillo de protoporfirina.
15. Porfobilinógeno
Uroporfirinógeno III Uroporfirinógeno I
Uroporfirinógeno I sintasa
Uroporfirinógeno III cosintasa
4 NH4
+
4 NH4
+
Uroporfirina III
excretada
Uroporfirina I
excretada
Uroporfirinógeno
descarboxilasa
4 CO2
Uroporfirinógeno
descarboxilasa
4 CO2
Coproporfirinógeno I
excretado
Coproporfirinógeno III
Coproporfirinógeno
oxidasa
4 H
2 CO2
Protoporfirinógeno III Protoporfirina III
Protoporfirinógeno III
oxidasa
6 H
HEMO
Ferroquelatasa
(hemo sintasa- mitocondrial)
Fe2+
85 % Hemoglobina
10 % Mioglobina
Hemoproteinas (citocromos)
M
16.
17. METABOLISMO DE LA BILIRRUBINA
Pigmento biliar de color amarillo
Se forma a partir del catabolismo del grupo hemo
Bilirrubina indirecta o no conjugada (insoluble en agua): 0,1 a 0,5 mg/dl
Bilirrubina directa o conjugada (soluble en agua): 0 – 0,3 mg/dl
Bilirrubina Total: 0,2 – 1 mg/ dl
18. CATABOLISMO
Destinos de la porción proteínica globina y del grupo
prostético hemo
macrófagos
Bazo
Colestasis extrahepática (obstrucción conductos biliares)
oxidasa
Transportado por la transferrina al hígado
donde se almacena como Fe 3+ o al
Bazo
Bilirrubina Indirecta
19.
20. SRE
Fase inicial SRE, ppalmente hígado, bazo, médula ósea
HEMOGLOBINA
Sist multienzimático hemo- oxigenasa del REL
-Convierte el C de los pirroles en CO, con apertura de los anillos
- Oxidación de Fe2+ a Fe3+
hemo- oxigenasa O2
NADPH + H+
NADP+
hemo- oxigenasa
O2
CO
Fe3+
BILIVERDINA (pigmento verde)
NADPH + H+
NADP+
Biliverdina
reductasa
BILIRRUBINA (pigmento amarillo-naranja, insoluble en agua)
Por cada gramo de Hb degradada origina 35 mg de billirubina
Sangre circulante HIGADO
Albumina
Billirubina conjugada
21.
22.
23. TRANSPORTE DE LA BILIRRUBINA
-Insoluble en agua
-Circula unida a proteínas plasmáticas (albúmina)
-Forma complejo macromolecular que no penetra en las células, ni
ultrafiltar a nivel glomérulos, no se excreta por orina
-Albúmina presenta sitio de alta y baja afinidad para unir bilirrubina
-A elevadas [bilirrubina], el exceso se une laxamente a los sitios de
baja densidad.
-Si la [bilirrubina] insoluble se eleva, se saturan los sitios de alta
afinidad y el exceso se transporta en unión lábil con proteínas, de las
que puede liberarse y difundir a las células.
-RN hiperbilirrubinemia…… eritroblastosis
-Compromiso SNC Kernicterus
25. ETAPA HEPÁTICA
- Se separa bilirrubina de albúmina y penetra en la célula por difusión
facilitada, mediada por transportador de membrana ( bilitranslocasa)
-En hepatocito se une a proteínas del citoplasma. Ligandina o prot Y
identificada como glutation –S – transferasa (GST)
-Bilirrubina es conjugada con moléculas polares y convertida en
producto soluble para ser excretada por bilis. Se produce en REL
2 UDP glucurónico
Bilirrubina Diglucurónido de bilirubina
Bilirubina-glucuronil transferasa BGT
Uridina difosfato-glucurónico (UDP-glucurónico)
26. BILIRRUBINA DIRECTA – INDIRECTA
Bilirrubina directa es glucurónido de bilirrubina, producto
soluble en agua formado en célula hepática.
Bilirrubina Indirecta, es el pigmento formado en el SRE, aún
no conjugado con glucuronato.
En plasma normal pequeña cantidad de bilirrubina, casi en su
totalidad indirecta, insoluble, transportada por albúmina.
VN= < 1,0 mg /dl . Valores superiores a 1,5 mg por dL
anormales
27. ETAPA INTESTINAL
En intestino el Glucurónido de bilirrubina es hidrolizado y sometido a
la acción reductora de sistemas enzimáticos de bacterias anaerobias
de la flora intestinal .
Se forma estercobilinógeno, compuesto incoloro que se elimina por
materia fecal.
En contacto con el aire el estercobilinógeno se oxida y se convierte en
estercobilina o urobilina, pigmento parduzco (coloración normal de
heces)
ETAPA ENTEROHEPÁTICA
Parte de los derivados de la reducción de bilirrubina se reabsorbe en
intestino y por via portal vuelve a hígado, donde se oxidan y
regeneran glucurónidos de bilirrubina y se excretan nuevamente con
la bilis hacia intestino.
29. ICTERICIA
Cuadros patológicos que alteran el catabolismo de Hemo.
Incremento en la concentración de bilirrubina en sangre, el pigmento
pasa a los tejidos y le da tinte amarillento.
Puede ocurrir por:
Hemólisis exagerada
Obstrucción de vías biliares
Insuficiencia funcional del parénquima hepático
En laboratorio se determina [Bilirrubina directa y total] en sangre
En orina de 24 hs urobilinoides
-Ictericia debido a alteración en el gen que codifica para bilirrubina
glucuronil transferasa “Síndrome de Crigler Najjar y de Gilbert”
30. Gilbert “La actividad de la glucuroniltransferasa es baja”
hiperbilirrubinemia
•podría comprometer defectos complejos en la absorción hepática
de la bilirrubina
• hiperbilirrubinemia no conjugada leve asintomática.
• hepatograma presenta resultados normales
•se diferencia de la hemólisis por la ausencia de anemia y
reticulocitosis.
•No requiere tratamiento
Crigler Najjar
enfermedad autosómica recesiva tipo I (completa).
•hiperbilirrubinemia grave y en general mueren debido a
encefalopatía hiperbilirrubinémica ( kernicterus ) hacia el año de vida
•trasplante de hígado
31. HIPERBILIRRUBINEMIA DEL RN
La piel y la esclerótica de los ojos del bebé amarillas. Esto se llama
ictericia.
•Incapacidad del hígado de captar y conjugar la bilirrubina
•↑ Bil indirecta o no conjugada
• Efectos tóxicos, afecta a las neuronas, dañando mitocondrias
(alteraciones del metabolismo energético y apoptosis), y de la
membrana plasmática ( alteración en el transporte de
neurotransmisores)
•Limites de riesgo > a 20mg/dL .
•Fototerapia (fotolabilidad de bilirrubina y que en presencia de
oxigeno se descompone en productos hidrosolubles de facil
eliminación
32. La ictericia grave del recién nacido puede ocurrir si el bebé tiene una afección que
aumente la cantidad de glóbulos rojos que necesitan ser reemplazados en el cuerpo,
como:
•Formas anormales de las células sanguíneas (como la anemia drepanocítica)
(enfermedad que se transmite de padres a hijos y en la cual los glóbulos rojos
presentan una forma semilunar anormal, hemoglobina S )
•Incompatibilidades del grupo sanguíneo entre el bebé y la madre (incompatibilidad
Rh)
•Sangrado por debajo del cuero cabelludo (cefalohematoma) causado por un parto
difícil
•Niveles más altos de glóbulos rojos, lo cual es más común en bebés pequeños para su
edad gestacional (PEG) y algunos gemelos
•Infección
•Falta de ciertas proteínas importantes, llamadas enzimas
Los factores que pueden dificultar la eliminación de la bilirrubina del cuerpo del bebé
también pueden llevar a que se presente ictericia más grave, incluyendo:medicinas,
Infecciones congénitas como rubéola, sífilis. Enfermedades que afectan el hígado o las
vías biliares, como la fibrosis quística o la hepatitis, Hipoxia Infecciones (sepsis
33. PORFIRIAS
↑ En la excreción de porfirinas o sus precursores
-Bloqueo en la síntesis de hemo
- Pueden tener origen hereditario o adquirida
-La producción de hemo esta deprimida, estimula la síntesis de la d
aminolevulinato sintasa.
-↑ Acumulación de intermediarios
-El hemo también se encuentra en la mioglobina, una proteína que
está en ciertos músculos.
-Las Porfirias de origen hereditario pueden ser eritropoyéticas o
hepáticas
39. Delta-ALA
Proteína producida por el hígado. Se busca un aumento en los niveles de Delta-ALA.
Muestra: orina de 24 hs
Preparación para el examen
Suspender medicación. Estos incluyen:
Penicilina (como antibiótico)
Barbitúricos (medicamentos para tratar la ansiedad)
Píldoras anticonceptivas
Griseofulvina (medicina para tratar infecciones micóticas)
VN: menos de 6 mg para una muestra de orina aleatoria. El rango normal es de 0 a 7
mg para una muestra de 24 horas.
Significado de los resultados anormales
Un nivel elevado de delta-ALA en la orina puede indicar:
Intoxicación con plomo
Porfiria (varios tipos)
Un nivel bajo se puede presentar con enfermedad hepática crónica (a largo plazo).
40. Examen de PBG en la orina
Muestra: Orina de 24 hs
Un resultado negativo se considera normal.
VN= menos de 4 miligramos por 24 horas.
Significado de los resultados anormales
Un aumento en los niveles de porfobilinógeno en la orina puede deberse a:
Hepatitis, Intoxicación con plomo, Cáncer del hígado, Porfiria (varios tipos)
Examen Porfirinas en sangre
Ayuno de 12 a 14 hs
Niveles totales de porfirinas: 16 a 60 mcg/dL
Nivel de coproporfirinas: < 2 mcg/dL
Nivel de protoporfirinas: 16 a 60 mcg/dL
Nivel de uroporfirinas: < 2 mcg/dL
Significado de los resultados anormales
Niveles elevados de coproporfirinas: Porf. eritropoyética congénita, Coproporfiria
hepática, Anemia sideroblástica, Porfiria variegata
Nivel elevado de protoporfirina:Anemia por enf crónica, Prot eritropoyética congénita
Aumento de eritropoyesis, Infección, Anemia ferropénica, Intoxicación con plomo
Anemia sideroblástica, Talasemia, Porfiria variegata
Nivel elevado de uroporfirina: Porfiria eritropoyética congénita
Porfiria cutánea tardía
41. PORFIRIAS HUMANAS HEREDITARIAS
PORFIRIAS HEPATICAS:
a) Porfiria aguda intermitente (PAI) autosómica dominante (62.1 %)
b) Porfiria cutánea tarda (PCT) P. hepatoeritropoyética (PHE) autosómica
dominante (31.5 %) forma homocigótica
c) Porfiria variegata (PV) autosómica dominante (1.6 %)
d) Coproporfiria hereditaria autosómica dominante (0.8 %)
PORFIRIAS ERITROPOYETICAS:
a) Porfiria eritropoyética congénita (PEC) autosómica recesiva (0.8%)
b) Protoporfiria eritropoyética (PPE) autosómica dominante (3.2%) Esta
es la clasificación que vamos a utilizar para las porfirias humanas.
42.
43.
44. SÍNTOMAS
La porfiria causa tres síntomas principales:
•Cólicos o dolor abdominal (únicamente en algunas formas de la enfermedad).
•Sensibilidad a la luz que causa erupciones, ampollas y cicatrización de la piel
(fotodermatitis).
•Problemas con los sistemas nervioso y muscular (convulsiones, alteraciones
mentales, daño neurológico).
Los ataques pueden ocurrir en forma súbita, generalmente con dolor de estómago
fuerte, seguido de vómito y estreñimiento.
El sol puede causar dolor, sensaciones de calor, ampollas, al igual que enrojecimiento e
hinchazón de la piel. Las ampollas sanan lentamente, a menudo con cicatrización o
cambios en el color de la piel, y pueden ser deformantes.
La orina se puede tornar de color rojo o marrón después de un ataque.
Otros síntomas incluyen:
Dolor muscular, Parálisis o debilidad muscular, Entumecimiento u hormigueo, Dolor en
brazos y piernas, Dolor de espalda, Cambios de personalidad
Los ataques algunas veces pueden ser mortales y producir: Presión arterial baja
Desequilibrios electrolíticos graves, Shock
45.
46.
47.
48.
49. Obstrucción vias biliares
Heces gris clara
No hay reabsorción a través del ciclo
Enterohepático y no se excretan urobilinoides
Por orina
Obstrucción vías biliares
50. Destrucción exagerada de GR
. ↑ Bilirrubina que pasa a plasma
.Bil conjugada con albúmina no filtra
por riñón.
51. Insuficiencia
funcional hepática
. Producción de Bil. Por SRE normal
. ↑ Bil indirecta en plasma
. Reflujo de bilis hacia capilares sanguineos
. ↑ Bil directa (glucurónido) en plasma
. Bil directa se elimina por riñón, orina
Oscura.
. ↓producción y excreción de bilis al
Intestino, reducción de urobilinoides
en heces y orina
60. La talasemia es un tipo de anemia del grupo de anemias hereditarias.
Causa una disminución de la síntesis de una o más de las
cadenas polipeptídicas de la Hb. Hay varios tipos genéticos, con cuadros
clínicos que van desde anomalías hematológicas difícilmente
detectables hasta anemia grave y cuadros de enfermedad terminal. La
hemoglobina del adulto, denominada Hemoglobina A está compuesta
por cuatro cadenas de polipéptidos: 2 cadenas α y 2 cadenas β. Hay
dos copias del gen que produce la hemoglobina α (HBA1 y HBA2), y cada
uno codifica una α-cadena, y ambos genes están localizados en
el cromosoma16. El gen que codificalas cadenas β (HBB) está localizado
en el cromosoma 11.
61. Talasemias
Causas Moleculares
Esta enfermedad está provocada por deleciones en uno o varios genes
de los que componen los grupos de la α-globina y la β-globina. Según
estas deleciones involucren más o menos genes el tipo de talasemia
será más o menos grave. Estas deleciones provocan la disminución en la
producción de cadenas α o β, según el lugar de la deleción; la escasez
de cadenas α se intenta compensar con un aumento de la producción
de cadenas β, y viceversa, lo que da lugar a la formación de
hemoglobinas inestables que provocan la destrucción de los glóbulos
rojos y por lo tanto anemia.
A su vez las deleciones parecen ser el resultado de entrecruzamientos
desequilibrados entre los segmentos duplicados presentes en la región
de la agrupación.
En el caso de las β-talasemias además de la deleción del gen de la β-
globina, también pueden darse por otras causas como:
•Mutaciones en el promotor que detienen o reducen su transcripción.
•Mutaciones en los sitios de corte y empalme (splicing) que impiden la
62. El defecto o deleción de un gen en la talasemia β causa una anemia
hemolítica que oscila entre leve y moderada sin síntoma alguno. La
deleción de dos genes ocasionan anemia más grave y la presencia de
síntomas: debilidad, fatiga, dificultad respiratoria. En las variantes más
graves, como la talasemia beta mayor, pueden aparecer ictericia, úlceras
cutáneas, cálculos biliares y agrandamiento del bazo (que en ocasiones
llega a ser enorme). La actividad excesiva de la médula ósea puede
causar el ensanchamiento y el agrandamiento de algunos huesos,
especialmente los de la cabeza y del rostro