Descargado 24 veces






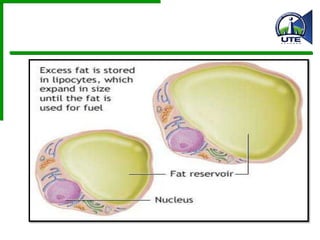
















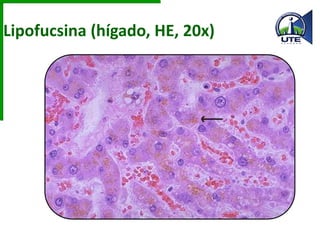







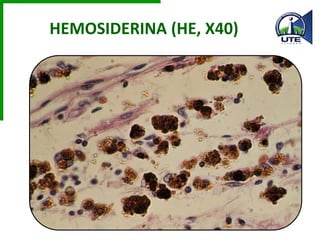


















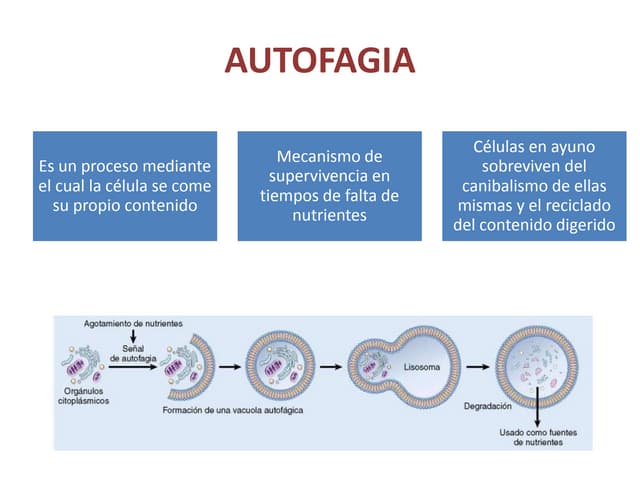
Este documento describe diferentes tipos de acumulaciones anormales que pueden ocurrir dentro de las células, incluyendo acumulaciones de grasas, colesterol, proteínas y pigmentos. Explica las causas de estas acumulaciones como defectos genéticos, toxinas como el alcohol, y trastornos metabólicos como la diabetes. También describe la morfología de estas acumulaciones y posibles mecanismos subyacentes.