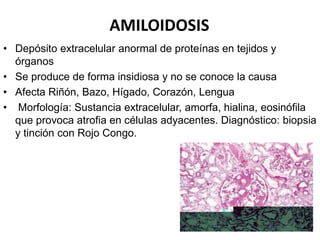
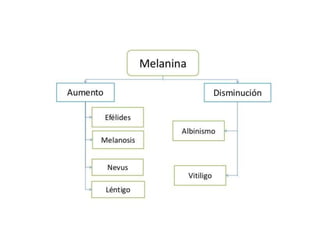
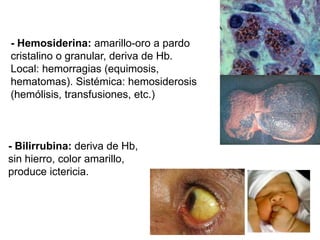
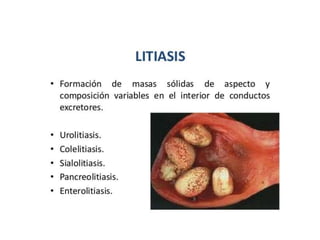
Este documento describe diferentes tipos de acumulaciones intracelulares, incluyendo acumulaciones de lípidos, proteínas, glucógeno y pigmentos. Explica las causas de estas acumulaciones, como trastornos metabólicos o la presencia de sustancias exógenas. Además, clasifica los tipos de acumulaciones y describe sus características morfológicas visibles al microscopio óptico y electrónico.