Descargar para leer sin conexión

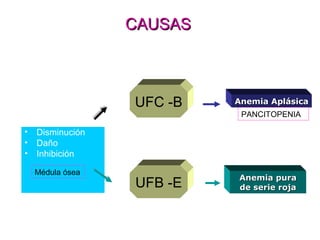
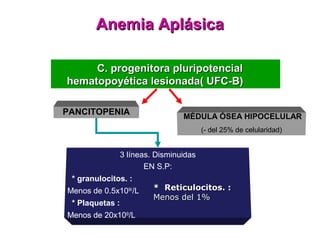
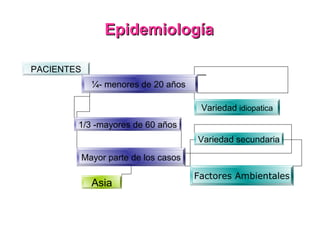
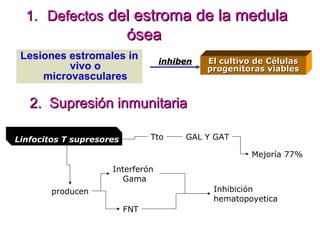


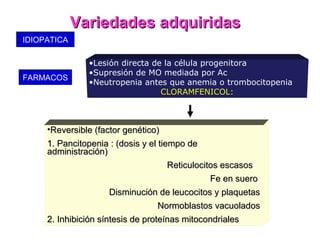
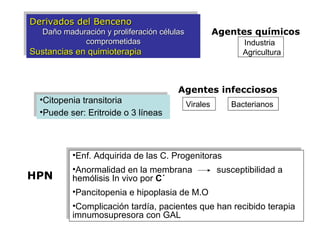
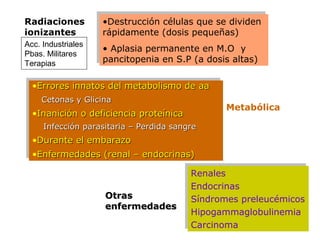
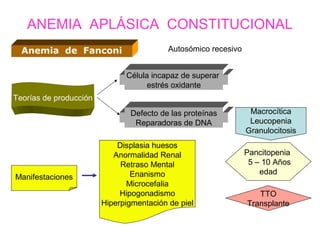

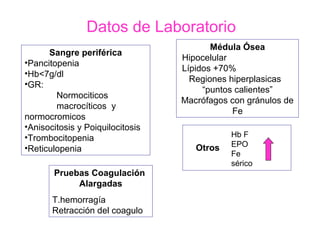
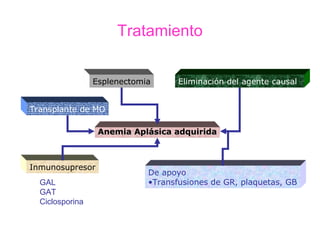



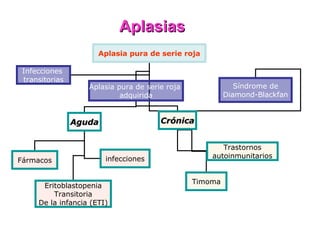
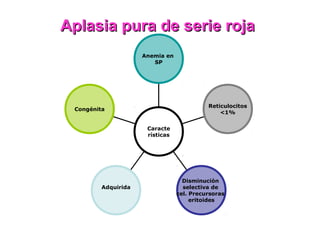
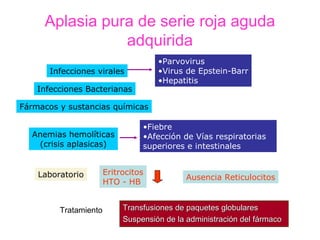
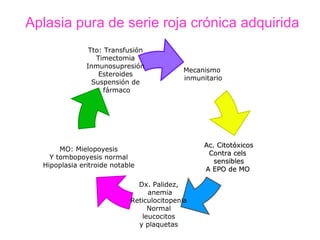
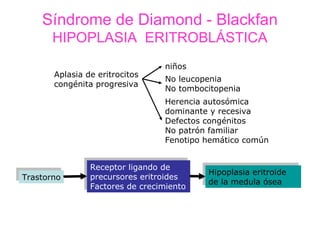






Este documento describe las causas y características de la anemia aplásica. La anemia aplásica se define por una disminución de las tres líneas sanguíneas (eritrocitos, leucocitos y plaquetas) debido a daño o inhibición de la médula ósea, lo que resulta en una médula ósea hipocelular. Puede ser adquirida por factores como medicamentos, radiación o infecciones virales, o puede ser constitucional como el síndrome de Fanconi. El tratamiento incluye transfusiones, inmun









































































