



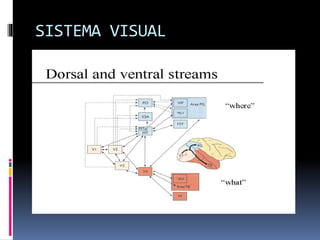

























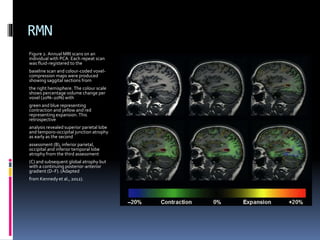


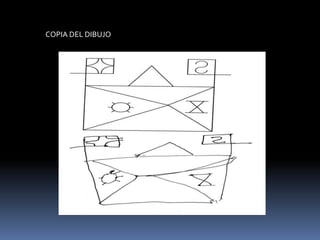
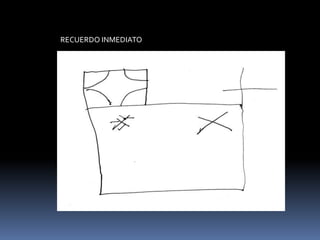











La atrofia cortical posterior (ACP) es un síndrome neurodegenerativo que afecta principalmente funciones visuoperceptivas y se caracteriza por atrofia en las áreas parieto-occipitales. Se presenta con trastornos visuales desproporcionados, preservando la memoria verbal en las etapas iniciales, y puede estar asociada con la enfermedad de Alzheimer. El diagnóstico se realiza postmortem y los estudios de imagen muestran atrofia focal asimétrica en las regiones afectadas.























































































