Descargado 305 veces


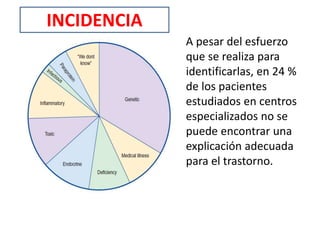
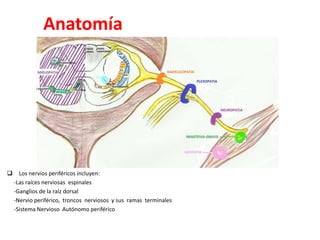
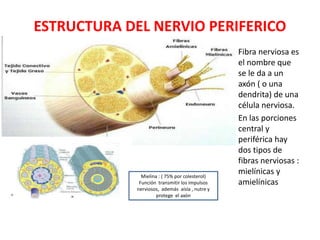
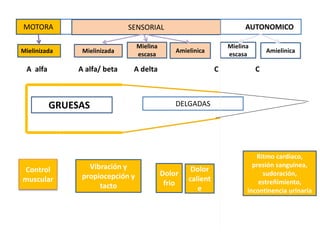




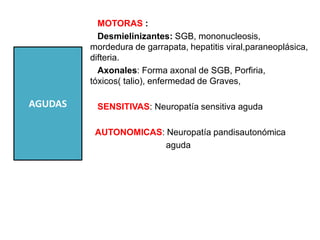



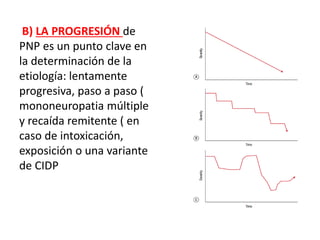
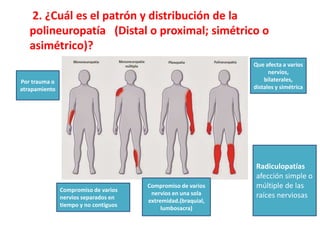


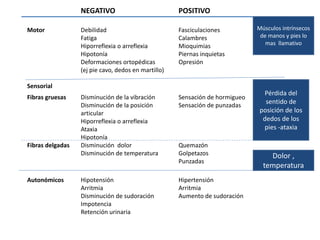


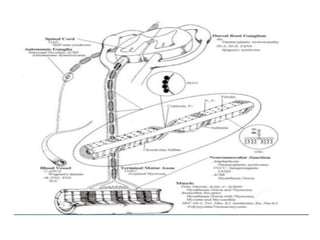
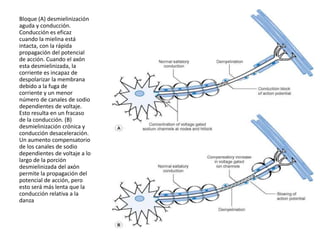



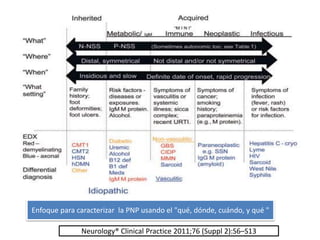

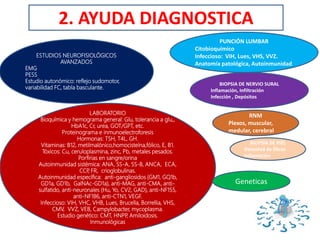
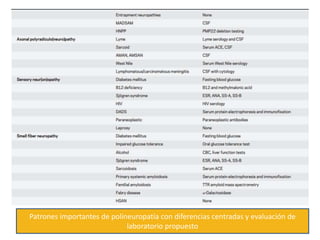



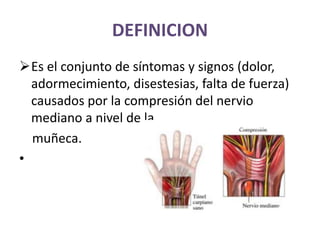

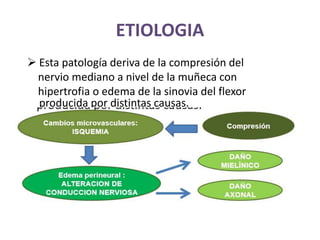

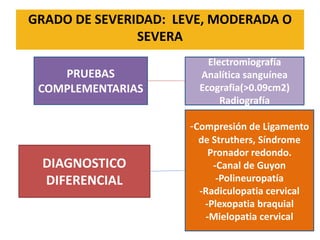

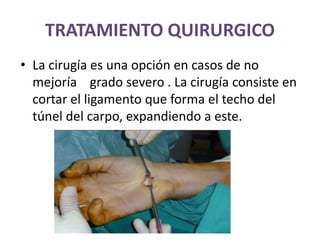

1) La polineuropatía es un trastorno de los nervios periféricos que causa cambios funcionales y patológicos y que compromete los nervios periféricos. 2) Existen varios tipos de polineuropatía dependiendo de si afecta a las fibras motoras, sensoriales o autonómicas, y si causa daño a la mielina o al axón. 3) El diagnóstico requiere identificar la causa evaluando factores como la distribución de los síntomas, antecedentes familiares, enfermed


































































































