

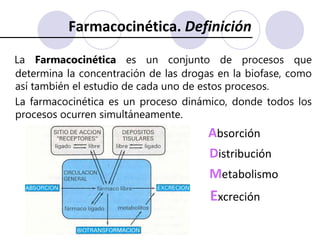
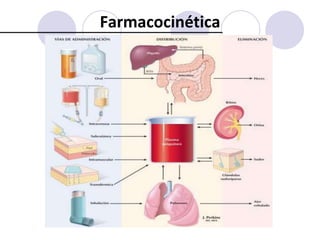



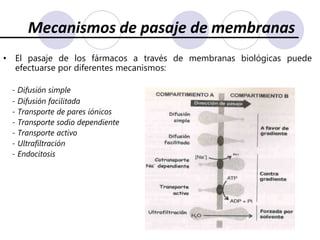




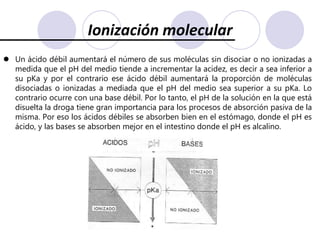


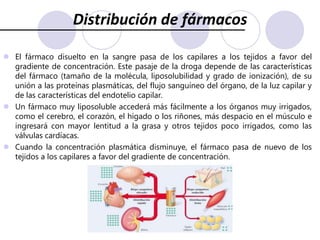
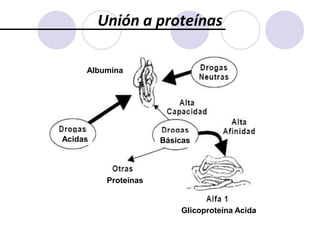
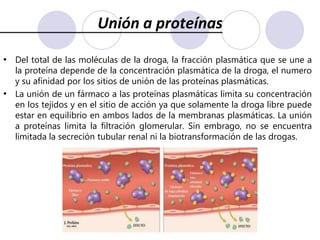





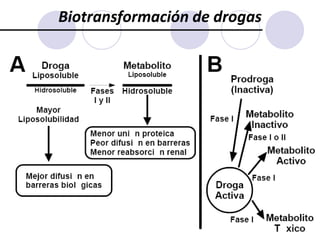


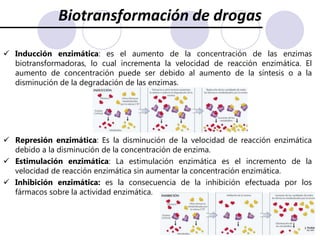
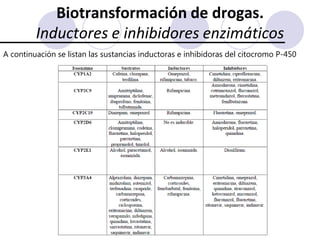

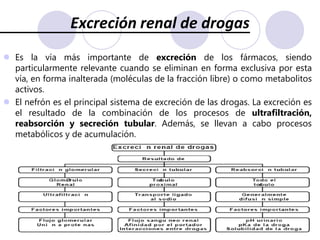






Este documento trata sobre la farmacocinética. Explica que la farmacocinética estudia los procesos de absorción, distribución, metabolismo y excreción de los fármacos en el organismo. Describe los diferentes mecanismos por los cuales los fármacos pueden pasar a través de las membranas biológicas, como la difusión simple, difusión facilitada y transporte activo. También analiza factores que afectan la absorción de los fármacos, como la liposolubilidad, tamaño molecular,































































