Descargado 238 veces
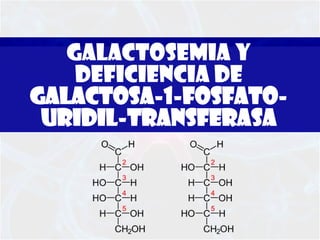











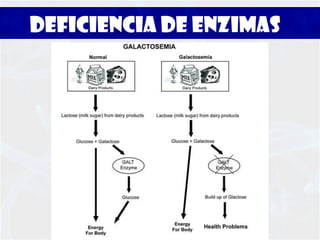








La galactosemia y deficiencia de galactosa-1-fosfato-uridil-transferasa son trastornos hereditarios del metabolismo de la galactosa que pueden causar daño a varios órganos como el hígado y el cerebro si no se tratan. La galactosemia clásica se debe a un defecto en la enzima galactosa-1-fosfato-uridil-transferasa y se diagnostica mediante pruebas en sangre y tratamiento con dieta libre de galactosa de por vida.










































































