Descargado 99 veces


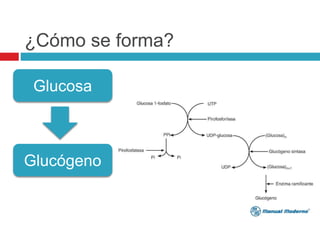














Este documento presenta información sobre las glucogenosis, un grupo de enfermedades hereditarias causadas por defectos en el metabolismo del glucógeno. Se describen 7 tipos principales de glucogenosis, incluyendo la enfermedad de Von Gierke, enfermedad de Pompe, enfermedad de Cori y enfermedad de McArdle, que se caracterizan por alteraciones en enzimas específicas involucradas en la formación, almacenamiento o degradación del glucógeno en el hígado y músculo.



































































