Descargar para leer sin conexión
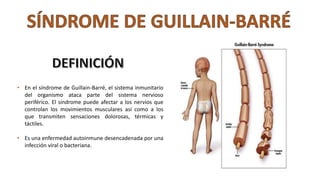


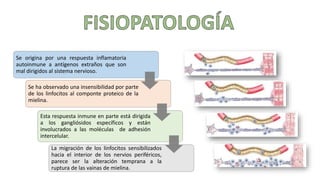
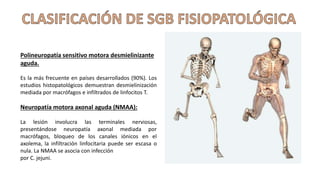
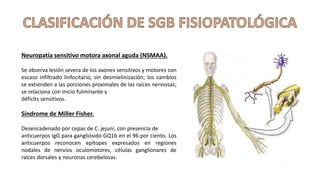
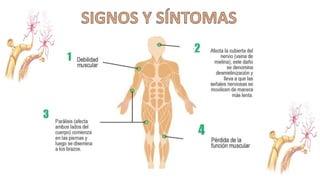
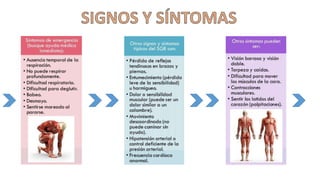
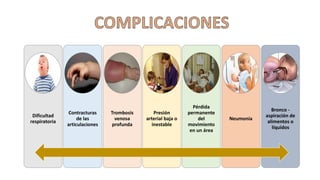
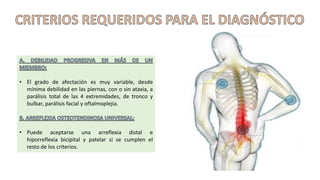

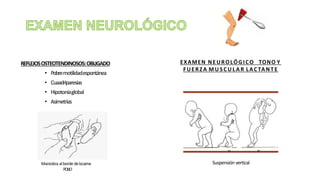
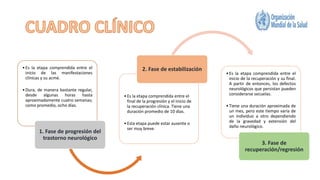










El síndrome de Guillain-Barré es una enfermedad autoinmune en la que el sistema inmunitario ataca y daña los nervios periféricos, causando debilidad muscular y, a veces, parálisis. Puede ser desencadenado por infecciones virales o bacterianas y afecta principalmente a adultos jóvenes. El tratamiento incluye terapias de apoyo e inmunoterapia para acortar la duración de la enfermedad y mejorar los síntomas.



































































