Descargado 179 veces






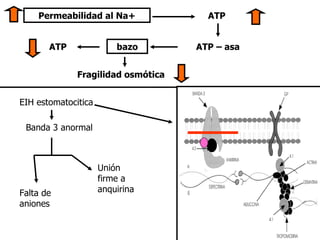

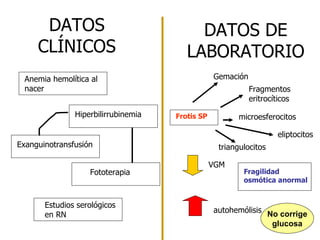

![ESTOMATOCITOSIS HEREDITARIA Anemia Hemolítica autosómica dominante K+ [ ] intracelular de cationes Células sobrehidratadas H 2 O intracelular E stomatocitos e Membrana anormalmente permeable Na+ Excede la capacidad de la bomba de cationes](https://image.slidesharecdn.com/ahdefectintrnsecos-100727102839-phpapp02/85/Ah-defect-intrinsecos-38-320.jpg)
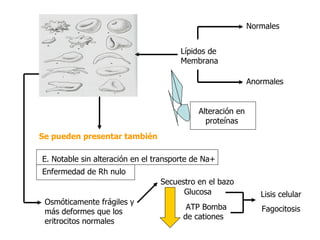

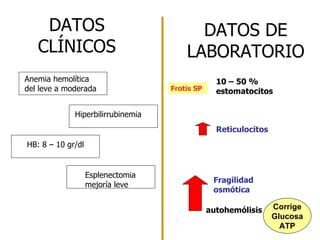

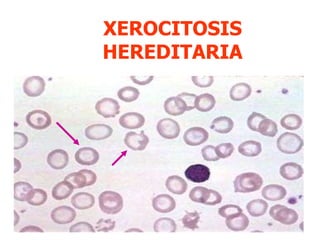
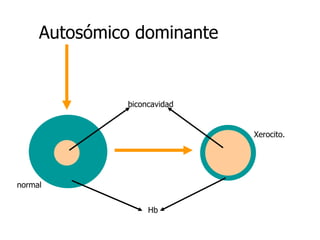
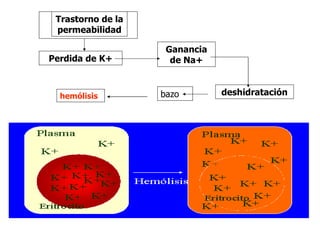
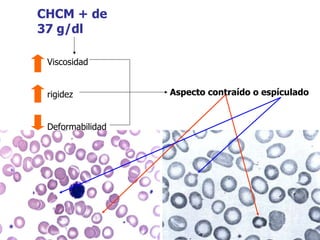

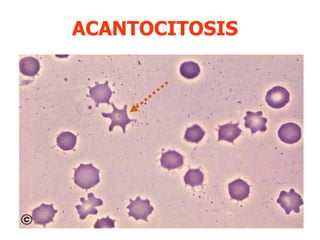
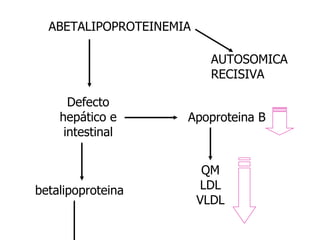
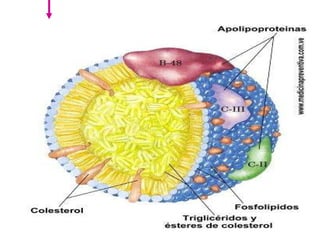
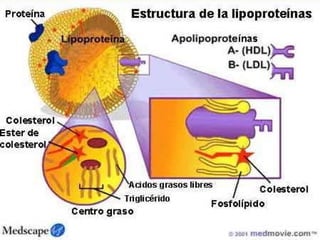
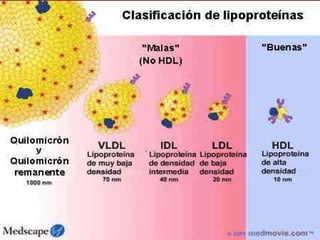
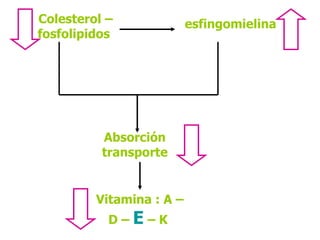
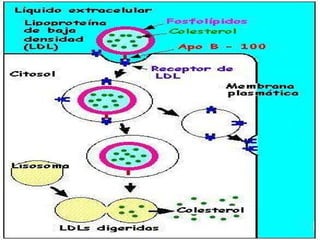
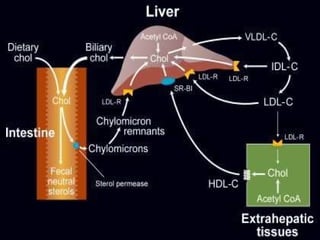
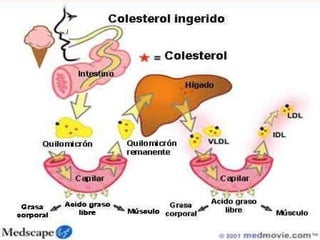
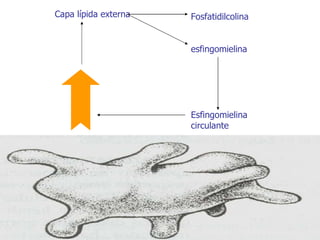


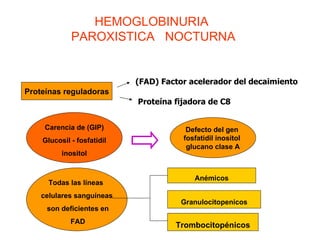
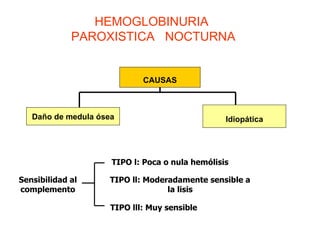
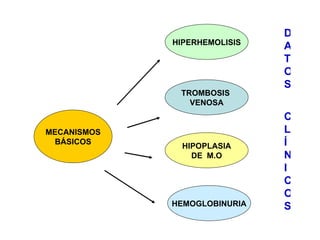
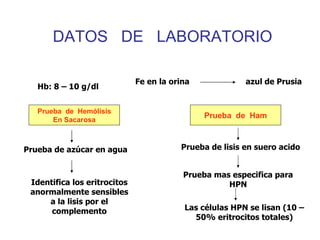
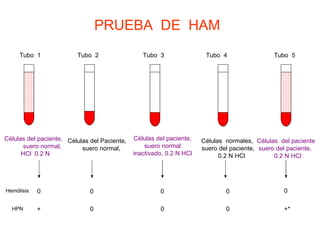
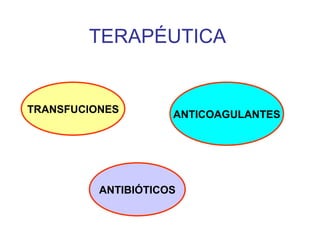


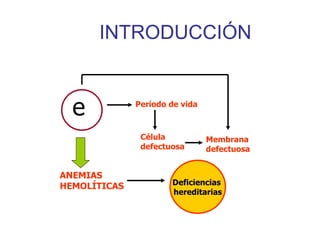
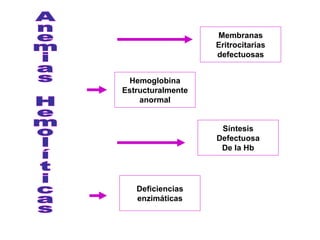
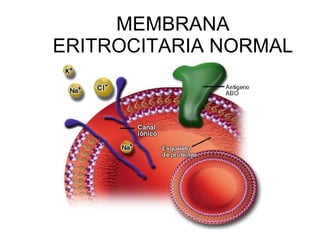
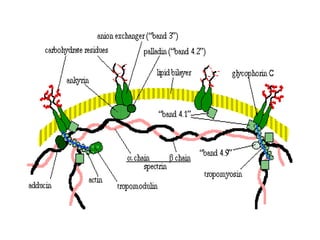
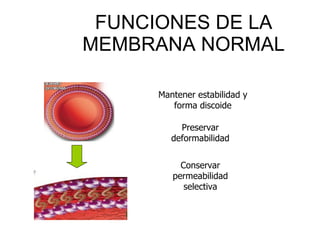
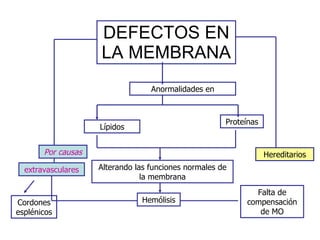
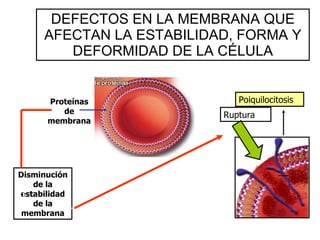
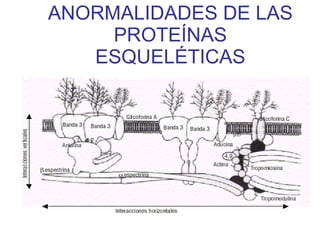
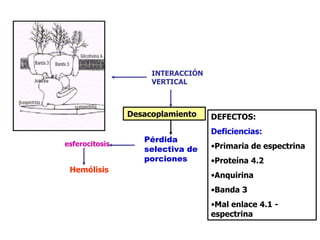
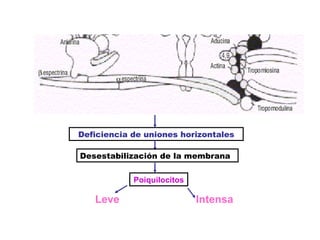
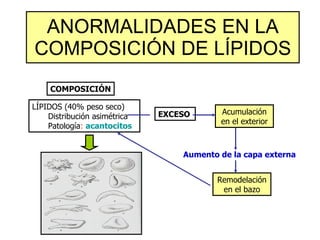
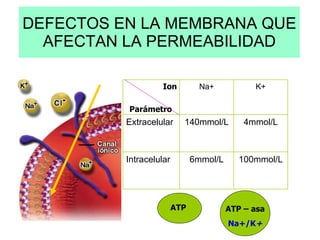
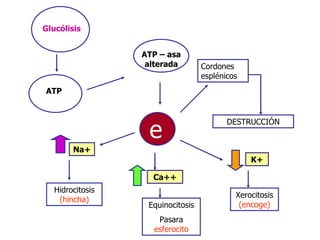
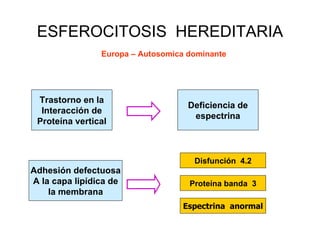
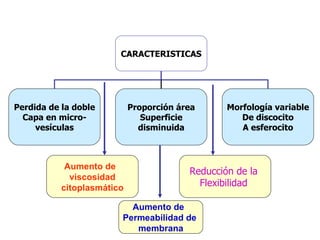
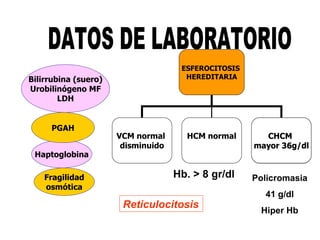
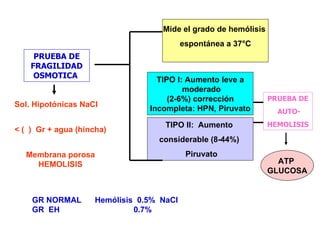
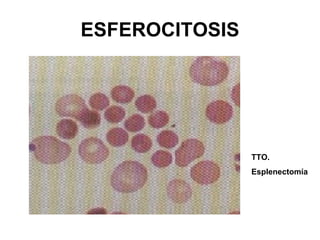
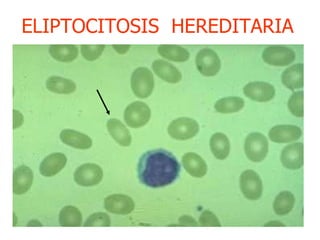
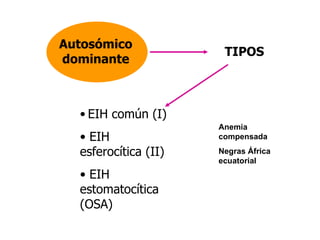
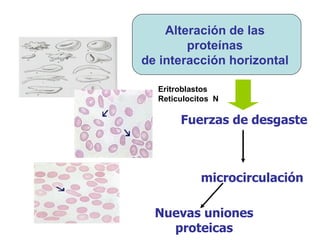
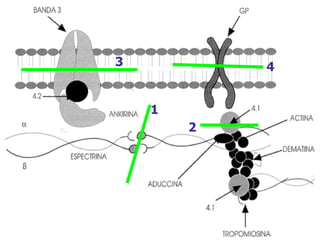
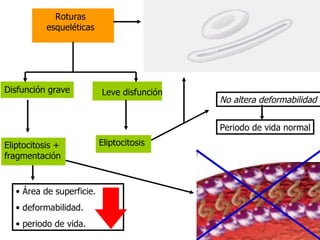
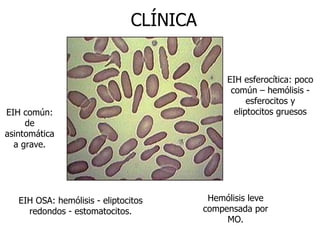
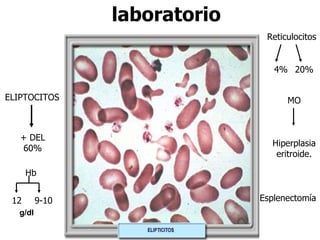
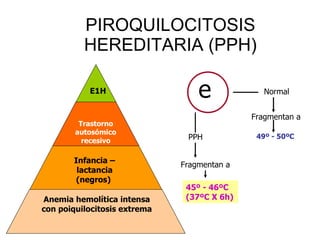
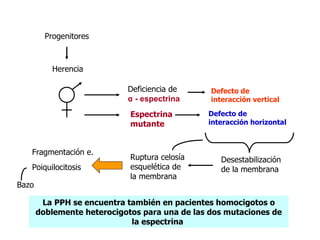
El documento describe diferentes tipos de anemias hemolíticas hereditarias causadas por defectos en la membrana eritrocitaria, incluyendo esferocitosis hereditaria, eliptocitosis hereditaria, piroquilocitosis hereditaria, estomatocitosis hereditaria y xerocitosis hereditaria. Estos trastornos se deben a mutaciones genéticas que afectan las proteínas de la membrana eritrocitaria y conducen a anormalidades en la forma, estabilidad, permeabilidad y funcionalidad de los eritrocitos.









































































