Descargado 412 veces






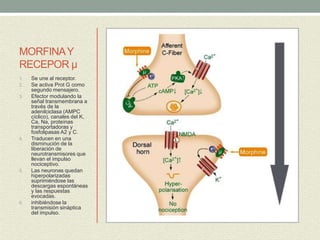



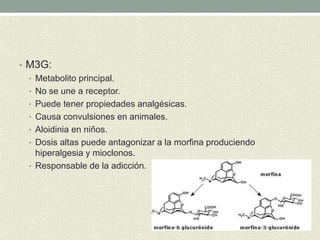







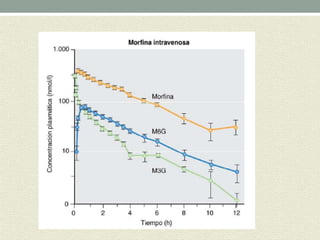


























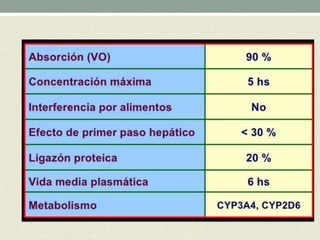

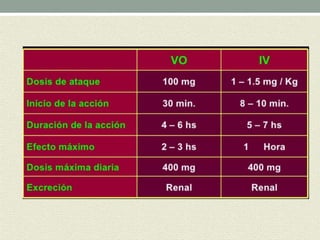





















Este documento describe las propiedades y usos de la morfina. La morfina es un potente analgésico extraído de la adormidera que actúa uniéndose a los receptores opioides en el sistema nervioso central. Tiene múltiples usos como analgésico, sedante preoperatorio y para el tratamiento del dolor asociado a enfermedades como infartos e cáncer. Sin embargo, también puede causar efectos adversos como depresión respiratoria, náuseas, estreñimiento y retención urinaria.



































































