Descargado 61 veces





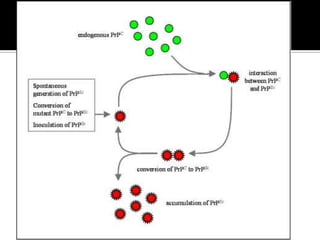







































































































Este documento describe las enfermedades priónicas o encefalopatías espongiformes transmisibles (EET), que son procesos neurodegenerativos caracterizados por la acumulación anormal de la proteína priónica. Las EET pueden presentarse de forma familiar, esporádica o adquirida, y su agente patógeno es la isoforma patológica de la proteína priónica llamada PrPSc. El documento analiza las características clínicas, anatomopatológicas y moleculares de las princip























































































