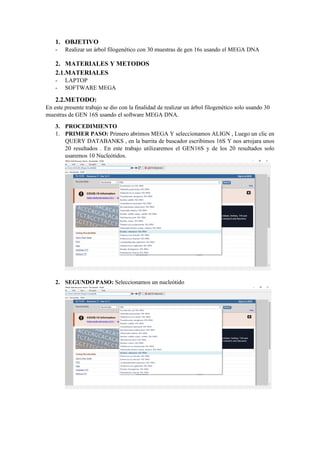
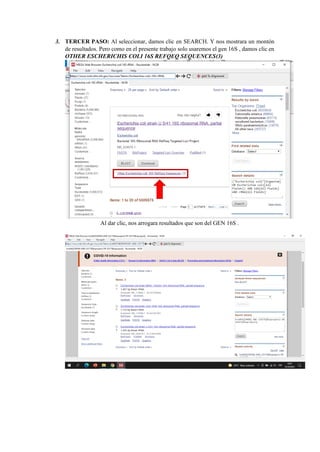
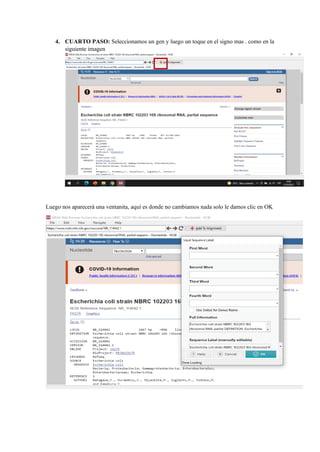
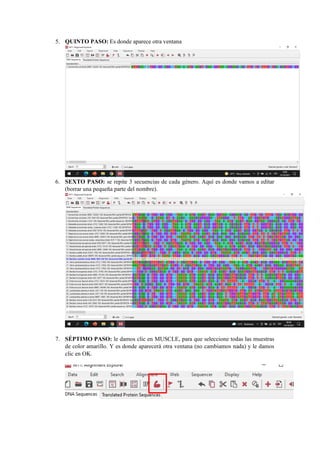
El documento describe el uso del software MEGA DNA para construir un árbol filogenético utilizando 30 muestras del gen 16S. Explica los pasos para recolectar las secuencias del gen 16S de una base de datos, alinear las secuencias y construir el árbol filogenético. Concluye que el software MEGA DNA es útil para este propósito y permite agrupar las especies de acuerdo a su categoría u otras características similares.