Descargado 786 veces







![• PKU: >1000 mol/L plasma y 500 mg/día
dieta.
• PKU no HPA: >120 mmol/L - <1000 mol/L
plasma.
• Variante PKU
Clasificación
inicial 1997
• PKU clásica: 250-350 mg/día dieta 300
mol/L (5mg/dL) plasma.
• PKU modera: 350-400mg/día
• PKU leve: 400 – 600mg/día
• HPA leve: [Phe] plasma < 600mmol/L
(10mg/dL) en una dieta normal.
Según
tolerancia de
Phe 1998](https://image.slidesharecdn.com/exposicionfenilcetonuria-131001204158-phpapp01/85/FENILCETONURIA-16-320.jpg)



![Métodos anticonceptivos.
Dieta restringida en Phe.
• 2-6 mg / dL.
Orientación nutricional continua.
Medición semanal o quincenal
de [Phe] plasmática.
Cuidado prenatal](https://image.slidesharecdn.com/exposicionfenilcetonuria-131001204158-phpapp01/85/FENILCETONURIA-28-320.jpg)


![Mitchell JJ. Phenylalanine Hydroxylase Deficiency. 2000 Jan 10 [Updated 2013
Jan 31]. In: Pagon RA, Adam MP, Bird TD, et al., editors. GeneReviews [Internet].
Seattle (WA): University of Washington, Seattle; 1993-2013. Disponible en
http://www.ncbi.nlm.nih.gov/books/NBK1504/
Pontificia Universidad Javeriana, Yeny Cuéllar, Homero Sáenz. Manejo
nutricional en pacientes con fenilcetonuria; Instituto de errores innatos del
metabolismo. Disponible en:
http://www.javeriana.edu.co/ieim/cartillas/fenilcetonuria.htm
Martínez-Pardo M, Bélanger-Quintana A, García Muñoz MJ, Desviat L, Pérez B,
Ugarte M. Protocolo de diagnóstico, tratamiento y seguimiento de las
hiperfenilalaninemias. Protocolos de la Asociación Española para el Estudio de los
Errores Congénitos del Metabolismo. Disponible en:
http://www.ae3com.eu/protocolos/protocolo4.pdf
Blau N, van Spronsen FJ, Levy L. Phenylketonuria. Lancet 2010; 376: 1417–27.
Champe P., Harvey R. y Ferrier D. Bioquímica. 4ta ed. Wolters Kluwer Health
España, S.A; 2008.](https://image.slidesharecdn.com/exposicionfenilcetonuria-131001204158-phpapp01/85/FENILCETONURIA-31-320.jpg)

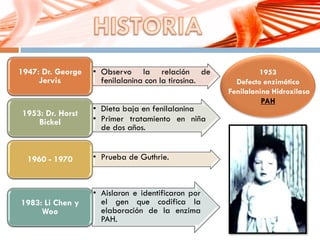
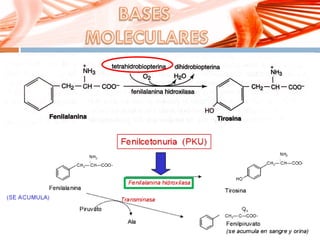
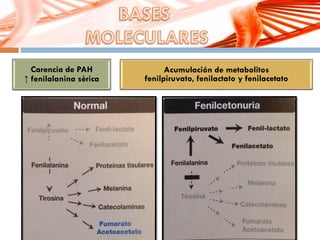
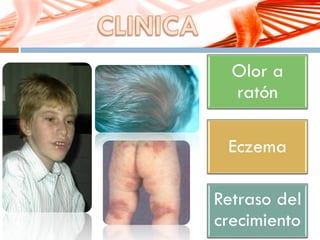
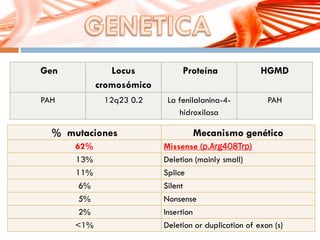
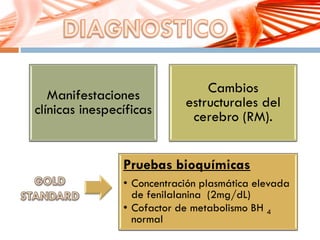
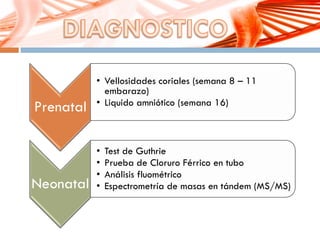
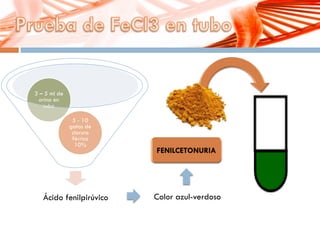
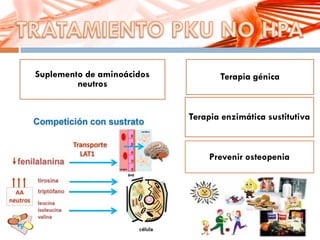
El documento describe la fenilcetonuria (PKU), un trastorno genético del metabolismo causado por un defecto en la enzima fenilalanina hidroxilasa. Esto provoca una acumulación tóxica de fenilalanina en la sangre que puede causar retraso mental si no se trata mediante una dieta baja en fenilalanina. Se explican los síntomas, las pruebas de diagnóstico, las opciones de tratamiento como la dieta y suplementos, y los aspectos genéticos y de seguimiento de la enfermed
































































