Descargado 101 veces




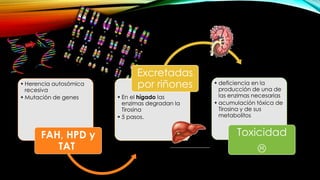
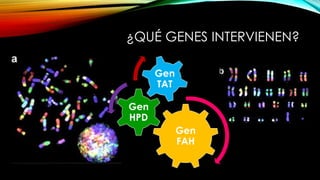
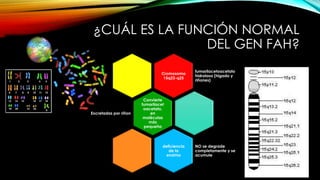
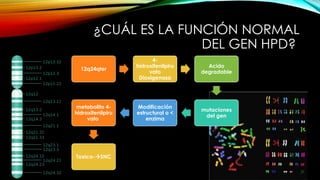
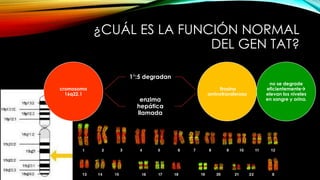
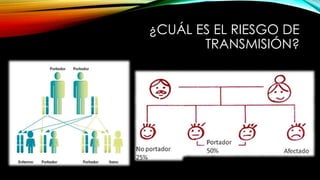

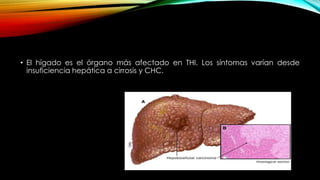
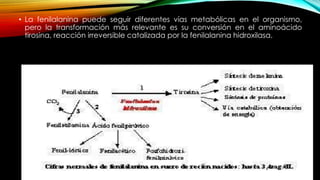


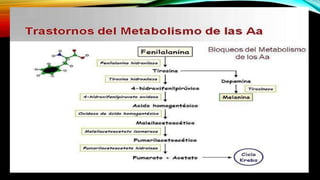


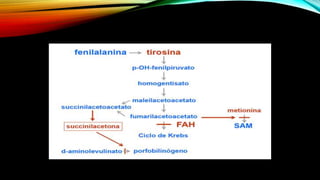




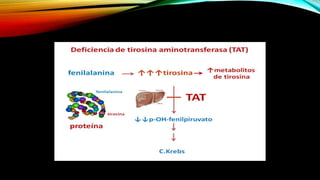


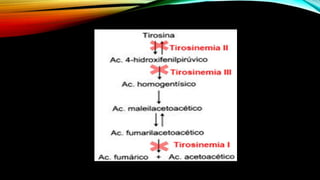
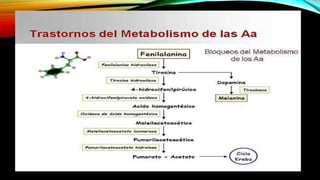





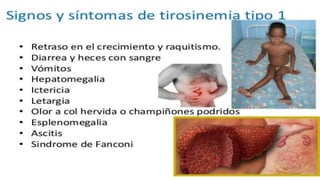

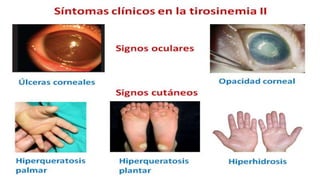
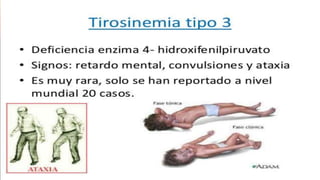

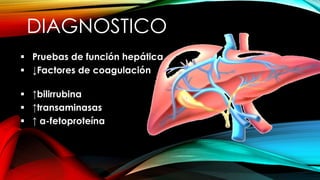




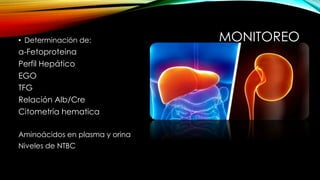


La tirosinemia es un trastorno metabólico congénito caracterizado por niveles elevados de tirosina en sangre, afectando principalmente el hígado y provocando complicaciones severas como insuficiencia hepática y riesgos de carcinogénesis. Existen tres tipos de tirosinemia, cada uno asociado a diferentes deficiencias enzimáticas y síntomas clínicos. El tratamiento incluye una dieta restringida y el uso de nitisinona, mejorando el pronóstico y reduciendo el riesgo de hepatocarcinoma.























































































