
Recomendados
Más contenido relacionado
Similar a POLICITEMIA VERA 1114-C.pptx.....mmmmmmm
Similar a POLICITEMIA VERA 1114-C.pptx.....mmmmmmm (20)
Último
Último (20)
POLICITEMIA VERA 1114-C.pptx.....mmmmmmm
- 2. EPIDEMIOLOGÍA El más frecuente de los trastornos mieloproliferativos crónicos. 2.5/100000 personas. Transmisión familiar infrecuente. Predomina después de la sexta década de la vida. Puede aparecer en cualquier edad, incluso en la niñez.
- 6. Presentación clínica Hemoglobina: Media: 18,4 g/dL Hematocrito: Media de 55% Esplenomegalia Prurito Trombosis arterial y venosa Hemorragia Hipertensión arterial Excoriación de la piel. Artritis gotosa(tofos) Ayalew Tefferi, M. (2023). Manifestaciones clínicas y diagnóstico de policitemia . UpTuDate, 35.
- 7. Prurito acuagénico: Desgranulación de los mastocitos: liberación de histamina y prostaglandinas. 1. Áreas involucradas: pecho, espalda y brazos. Eritromelalgia: Dolor ardiente en zonas acras acompañado de eritema. 1. Complicaciones trombóticas microvasculares patognomónicas en la PV. Williams. Manual de Hematología, 10e CAPÍTULO 42: Policitemia verdadera
- 8. • Trombosis: (EVC, infarto de miocardio, TVP, EP). Primera causa de mortalidad por PV 1. Estado de hipercoagulabilidad: Aumento de la viscosidad sanguínea. 2. Se debe sospechar PV en pacientes con síndrome de Budd –Chiari o trombosis de la vena porta o mesentérica, particularmente mujeres < 45 años.
- 9. • Alteraciones visuales transitorias: amaurosis fugaz, escotomas centellantes. Reducción del flujo sanguíneo coroideo y retiniano. Ceguera ocular transitoria Aumento del hematocrito
- 10. • Síntomas gastrointestinales: 1. Malestar epigástrico, antecedentes de úlcera-péptica. 2. Alteraciones en el flujo sanguíneo de la mucosa gástrica debido a la alteración de la viscosidad de la sangre o por la liberación de histamina. Ayalew Tefferi, M. (2023). Manifestaciones clínicas y diagnóstico de policitemia . UpTuDate, 35. Harrison. Principios de Medicina Interna, 21e, Policitemia vera y otras neoplasias mieloproliferativas. Elevada tasa de recambio de células hematopoyéticas. Hiperuricemia 1. Cálculos de acido úrico. 2. Gota (tofos)
- 11. Diagnóstico Biometría hemáticas completa • Serie roja: Aumento de numero de hematíes, Hb y Hto con VCM disminuido • Serie blanca: Aumento de leucocitos (Neutrofilos) • Serie megacariocitica: Aumento de plaquetas (Alt función) • Niveles de eritropoyetina baja Pruebas para las mutaciones JAK2V617F (Exón 14), JAK2 (Exón 12), CARL y LNK Estudio de médula ósea (Aspirado, cariotipo y biopsia) Díez, F. (2019). Manual de hematología. México: AMIR Liesveld, J. (s/f). Policitemia vera. Manual MSD versión para profesionales. Recuperado el 11 de enero de 2024, de https://www.msdmanuals.com/es/professional/hematolog%C3%ADa-y-oncolog%C3%ADa/trastornos-mieloproliferativos/policitemia-vera
- 12. Vista de Policitemia vera: presentación clínica, diagnóstico y nuevos abordajes terapéuticos. (s/f). Edu.co. Recuperado el 11 de enero de 2024, de https://revistasum.umanizales.edu.co/ojs/index.php/archivosmedicina/article/view/2681/3651 Se requiere la presencia de 3 criterios mayores o la presencia de los primeros criterios mayores junto el criterio menor
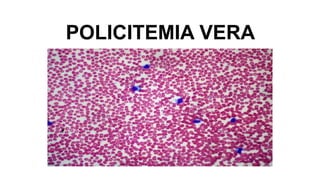
- 13. Sangre periférica •Eritrocitos absoluta •Leucocitosis moderada •Neutrófilos con poco mietamielocitos •Eosinófilo y basófilo positivo Médula ósea •Hipercelular con panhiperplasia •Los megacariocitos pueden ser anormales y tamaño y forma
- 14. Diagnóstico diferencial Neoplasias mieloproliferativas Eritrocitosis secundaria (Poliglobina) Enfermedad de von Willebrand Guías Prácticas Clínicas PARA DIAGNÓSTICO Y TRATAMIENTO DE NEOPLASIAS MIELOPROLIFERATIVAS CRÓNICAS CLÁSICAS BCR-ABL NEGATIVAS Mielofibrosis primaria Policitemia vera Trombocitemia esencial. (s/f). Sochihem.cl. Recuperado el 11 de enero de 2024, de https://www.sochihem.cl/bases/arch1689.pdf Liesveld, J. (s/f). Policitemia vera. Manual MSD versión para profesionales. Recuperado el 11 de enero de 2024, de https://www.msdmanuals.com/es/professional/hematolog%C3%ADa-y-oncolog%C3%ADa/trastornos-mieloproliferativos/policitemia-vera
- 15. Tratamiento Considerar tratamiento en fase pletórica y de gasto por separado Alopurinol para cifras altas de ácido úrico Medidas generales para aliviar síntomas Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera Lamielosupresióna diario con hidroxiurea, como terapia inicial (1500mg/dia) y como tratamiento a largo plazo (500-2000mg/dia) para mantener la Hb y los recuentos de neutrófilos y plaquetas en cifras normales Flebotomía algunos pacientes la requieren para mantener la Hb y plaquetas en rango IFN pegilado,ruxolitinib alternativa a la hidroxiurea Aspirinaen dosis de 80 a 100mgdia a todo paciente sin antecedente de sangrado o intolerancia gástrica importante y cuando el recuento plaquetario <1000x10^9 (para prevención de eventos trombóticos)
- 16. Riesgo alto Pacientes con trombosis o ataques isquémicos transitorios y sangrado previo atribuibles a PV Riesgo intermedio Pacientes > 60 años Riesgo bajo Pacientes < 60 años y sin antecedentes de trombosis Considerar fx riesgo adicionales: HTA no controlada, tabaquismo, DM, recuento alto de leucocitos Terapia mielosupresura Tratamiento Fase pletórica Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 17. Mielosupresión Disminuye los recuentos sanguíneos, el riesgo de eventos vasculares y los síntomas, aumenta la sensación general de bienestar. Hidroxiurea • Dosis desde 500 hasta 2500 mg al día • Tratamiento de primera línea • Es la terapia menos cara • Su efecto supresor es de corta duración - Terapia continua - seguro: cuando hay supresión excesiva los recuentos sanguíneos se recuperan en días • No daña al ADN no causa transformación leucémica • Px con PV propensos a cáncer cutáneos de células basales o de células escamosas aumenta mucho la frecuencia y agresividad Tratamiento Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 18. Interferón pegilado • Aprobación FDA ¿? • Muy caro • Con la duración prolongada este fue más eficaz que la hidroxiurea para normalizar los recuentos sanguíneos y reducir la carga de mutación impulsora Ruxolitinib • Recomendado en pacientes que no toleran la hidroxiurea • Pacientes con esplenomegalia importante y síntomas de fatiga y sudores nocturnos • Muy costoso Busulfan • Solo en casos seleccionados Tratamiento Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 19. Flebotomía • Se usa mejor de manera conjunta con mielosupresión • Recomendado para la enfermedad de bajo riesgo • Inicial: de 450 a 500 mL alrededor de cada 4 días hasta alcanzar un Hto diana • Contribuye a deficiencia de Fe+ • Por si sola se asocia a una incidencia mas alta de eventos trombóticos Tratamiento Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 20. Eritroaferesis • Alternativa terapéutica a la flebotomía. • Objetivo: reducir el volumen sanguíneo y el recuento de GR. • Eritroaferesis ofrece un método mas eficiente en el agotamiento de GR en comparación con la flebotomía, reduciendo potencialmente el numero de procedimientos de tratamiento necesarios para la inducción de px con PV, así como el intervalo entre procedimientos. Tratamiento Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 21. • Ocurre por lo general después de 10 años. • Los requerimientos de mielosupresión y flebotomía disminuyen y cesan. • Eritrocitos en lagrima, nucleados. • Anemia progresiva. • Leucocitosis o leucopenia. • Trombocitosis o trombopenia. • Leucocitos inmaduros. • Esplenomegalia progresiva. • Síntomas sistémicos (fatiga, sudores nocturnos, dolor óseo). Trasplante alogénico de células madre: única terapia curativa Tratamiento Fase de gasto Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 22. Pacientes no elegibles para trasplante • Inhibidores JAK2 • Mielosupresión suave con dosis bajas de hidroxiurea en presencia de leucocitosis y trombocitosis • Transfusión de eritrocitos o agentes estimulantes de eritropoyesis • Talidomida • Andrógenos • Terapias experimentales • Medidas generales y control de dolor • Esplenectomía para bazo muy grande Tratamiento Williams. Manual de hematología, 10e . CAPITULO 42: Policitemia verdadera
- 23. Complicaciones >índice de mortalidad la trombosis. Hemorragias. Eventos cardiovasculares. Daño a órganos blancos. Mielofibrosis. Leucemias. Vista de Policitemia vera: presentación clínica, diagnóstico y nuevos abordajes terapéuticos. (s/f). Edu.co. Recuperado el 11 de enero de 2024, de https://revistasum.umanizales.edu.co/ojs/index.php/archivosmedicina/article/view/2681/3651
- 24. Pronóstico Supervivencia mediana sin tratamiento es de 2 años. Supervivencia mediana con tratamiento de 10-15 años. Algunos estudios citan una mediana de supervivencia de 24 años, pero muchos pacientes viven mucho más tiempo, incluso cuando se desarrolla mielofibrosis. Díez, F. (2019). Manual de hematología. México: AMIR Liesveld, J. (s/f). Policitemia vera. Manual MSD versión para profesionales. Recuperado el 11 de enero de 2024, de https://www.msdmanuals.com/es/professional/hematolog%C3%ADa-y-oncolog%C3%ADa/trastornos-mieloproliferativos/policitemia-vera
- 26. Ficha de identificación. Masculino de 33 años de edad AHF. Madre fallecida de 70 años por ERC. Se desconoce padecimientos de línea paterna.
- 27. Padecimiento actual DM, HAS de 15 y 10 años de evolución respectivamente. Fumador activo a razón de 2 cajetillas semanales. Cefalea, bochornos, hormigueo de manos acompañado en ocasiones de dolor quemante. Presencia de cólico aparentemente biliar.
- 29. Exámenes complementarios: • Química sanguínea: 1. Hemoglobina:18.0 g/dl 2. Hematocrito: 54% 3. Plaquetas: 250,000 • USG Abdominal: 1. Hepatoesplenomegalia • Biopsia MO: Celularidad aumentada, con hiperplasia eritroide, granulocitica megacariocítica (panmielosis). • Mutación JAK2 V617F (+)
- 30. Criterios diagnósticos de la OMS Policitemia vera Criterios principales Criterio menor Hemoglobina >16.5 g/dl ò Hematocrito >49% ò Aumento de la masa de glóbulos rojos Nivel de eritropoyetina sérica subnormal. Biopsia de medula ósea que muestra hipercelularidad con crecimiento de los tres linajes: Proliferación eritroide, granulocítica y megacariocítica. El diagnóstico de PV requiere cumplir los 3 criterios principales o los 2 primeros criterios mayores y el criterio menor. Presencia de JAK2V617F o JAK2 mutación del exón 12
- 31. Tratamiento • Flebotomías cada 45 días • Mantiene: - Hto entre 42% -45%. - Plaquetas normales. • Esplenomegalia lentamente progresiva • 4 años de evolución: se agrega Hydrea (ajuste de dosis desde 500 mg trisemanal a 1gr/día) Hematocrito H: 42 a 52 % Buscar valores de GB
- 32. • Dolor abdominal intenso en hipocondrio izquierdo, asociado a sensación febril y sudoración nocturna • Bazo: 14 cm BCR • Biopsia de MO: celularidad conservada, con hiperplasia megacariocítica. • Esplenectomía: bazo 1500 gr, y trombosis de vena porta. 10 años de evolución (43 años)
- 33. 20 años de evolución (53 años) Biopsia de MO Moderadamente hipercelular. Aumento de áreas de fibrosis. Aumento de megacariocitos irregulares Disminución de la serie eritroide y mieloide, con caracteres de neoplasia mieloproliferativa, concordante con fase “post- policitemica”
- 34. 27 años de evolución (61 años) Asintomático Hepatomegalia progresiva. Hto. 35% estable. Anisopiquilocitosis severa con presencia de dacriocitos y eliptocitos, corpúsculos de Howell Jolly (+) GB aumentan: 15-20x10^3/ul Plaquetas >1000x10^3/ul LDH elevada hasta 3000 mg/dl Blastos 4%
- 38. • Complicaciones Hepatomegalia gigante Trombosis de la porta Evolución a mielofibrosis
- 39. Bibliografía Díez, F. (2019). Manual de hematología. México: AMIR Guías Prácticas Clínicas PARA DIAGNÓSTICO Y TRATAMIENTO DE NEOPLASIAS MIELOPROLIFERATIVAS CRÓNICAS CLÁSICAS BCR-ABL NEGATIVAS Mielofibrosis primaria Policitemia vera Trombocitemia esencial. (s/f). Sochihem.cl. Recuperado el 11 de enero de 2024, de https://www.sochihem.cl/bases/arch1689.pdf Liesveld, J. (s/f). Policitemia vera. Manual MSD versión para profesionales. Recuperado el 11 de enero de 2024, de https://www.msdmanuals.com/es/professional/hematolog%C3%ADa-y- oncolog%C3%ADa/trastornos-mieloproliferativos/policitemia-vera Vista de Policitemia vera: presentación clínica, diagnóstico y nuevos abordajes terapéuticos. (s/f). Edu.co. Recuperado el 11 de enero de 2024, de https://revistasum.umanizales.edu.co/ojs/index.php/archivosmedicina/article/view/2681/36 51
- 40. Bibliografía