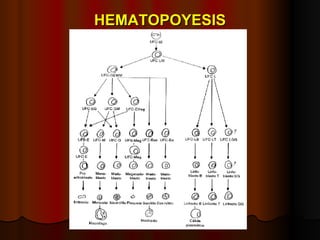
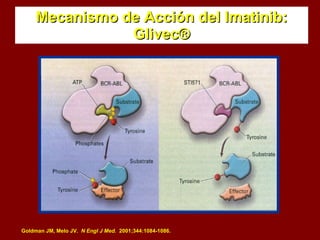
El documento describe los síndromes mieloproliferativos crónicos, incluyendo la leucemia mieloide crónica y la policitemia vera. La leucemia mieloide crónica se caracteriza por la presencia del cromosoma Filadelfia, resultando de una translocación entre los cromosomas 9 y 22. La policitemia vera es el resultado de la proliferación anómala de células pluripotenciales que son insensibles a la eritropoyetina. Ambas enfermedades cursan con hipercelularidad medular