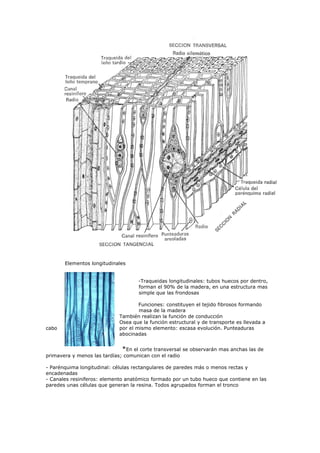
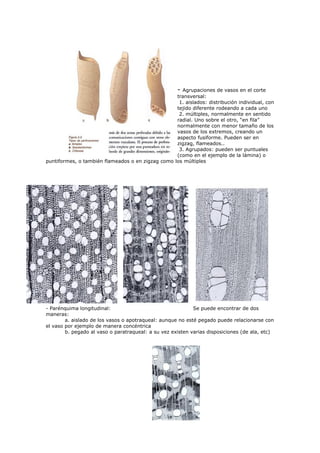
El documento detalla métodos científicos aplicados a la conservación de bienes culturales, incluyendo técnicas de análisis destructivas y no destructivas, para entender la composición y estado de conservación de las obras. También discute la interacción de radiaciones electromagnéticas con la materia y su aplicación en técnicas de examen, como fluorescencia ultravioleta y fotografía infrarroja, destacando su utilidad en la identificación de materiales y la evaluación del estado de conservación. Se enfatiza la importancia de seleccionar métodos adecuados en función de los objetivos del análisis y las características del material.






































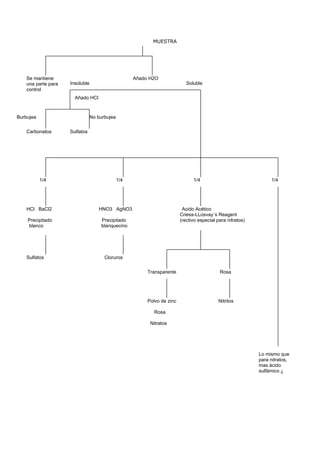
![Fe2O3 + HCl.............................FeCl3 + H2O
Óxido insoluble ácido concentrado........................cloruro
soluble
amarillento
- Emplearemos varios reactivos:
4-
a. ferrocianuro potásico K4 [Fe (CN)6]
Fe3+ + K4 [Fe (CN)6]4 - ..........................Fe3+4 [Fe (CN)6]4 -3
(prescindimos del K,
q es acompañante,..................................... Ferrocianuro férrico
(azul de Prusia)
nos interesa el anion ferrocianuro)
b. Este anión, en un medio alcalino, cambia de color hacia el amarillo. Por ello no se da este
pigmento en pintura mural, ya que viraría el color por la basicidad del sustrato.
Fe3+4 [Fe (CN)6]4 -3 + Na (OH)................Fe (OH)3
Ferrocianuro férrico Hidróxido sódico.................. hidróxido férrico
Amarillo
c. Con otro reactivo, el sulfocianuro potásico, obtenemos sulfocianuro férrico, de color pardo
rojizo
Fe3 + K (SCN)........................Fe (SCN)3-6
El Cobre
Cu0 Cu+ (cuproso) Cu2+
(cúprico)
Dos compuestos del cobre, la malaquita y la azurita, presentan carbonato de cobre e
hidróxido de cobre en diferentes proporciones (malaquita 1:1; azurita 2:1)
1CuCO3.1Cu (OH)2...........................malaquita
2CuCO3. 1Cu (OH)2 .........................azurita
- Ambas son insolubles, por lo que las atacaremos con ácido clorhidrico:
CuCO3. Cu(OH)2 + HCl....................CuCl2 + etc *Cu+ + Cl-2
Cloruro soluble
- Reacciona con ferrocianuro potásico
CuCl2 + K [Fe (CN)6]3.......................... Cu2Fe (CN)2 *Cu2+ + Fe
(CN)4 -6
Ferrocianuro de cobre
Viscoso, rojizo
- O también con hidróxido amónico](https://image.slidesharecdn.com/quimicasabe-100905123349-phpapp01/85/Quimicasabe-45-320.jpg)








































































![PPT_-_TRABAJO_DE_CIENCIA[1] abi1.pptxwdw](https://cdn.slidesharecdn.com/ss_thumbnails/ppt-trabajodeciencia1abi1-250322231844-c089a266-thumbnail.jpg?width=640&height=640&fit=bounds)






