Descargado 771 veces


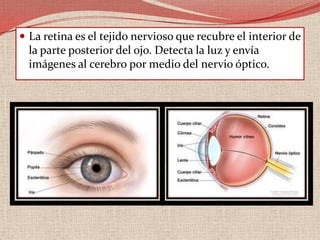
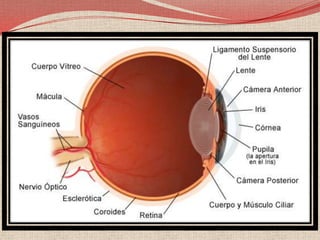
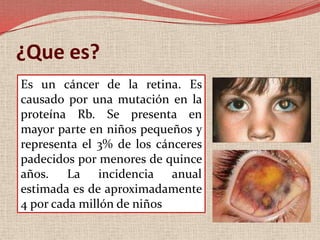




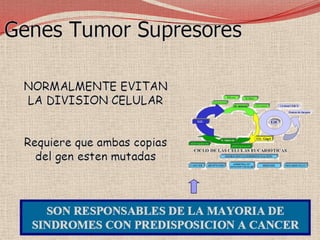
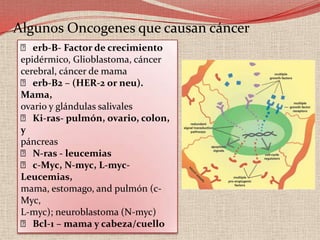
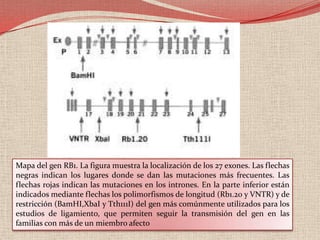


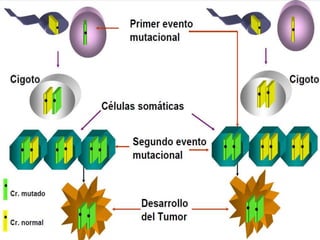
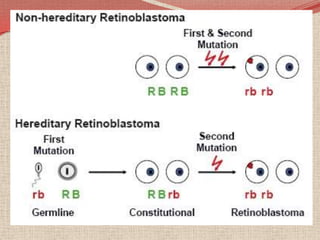
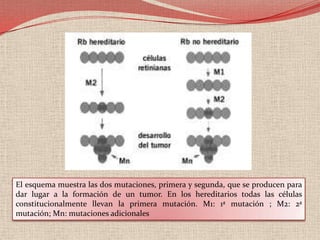

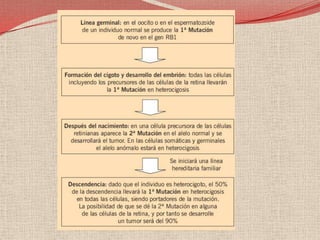





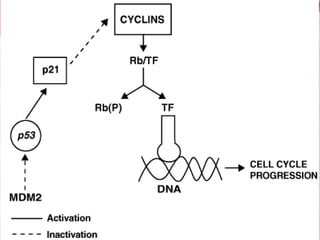

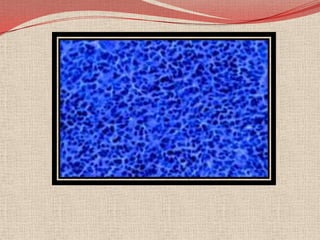
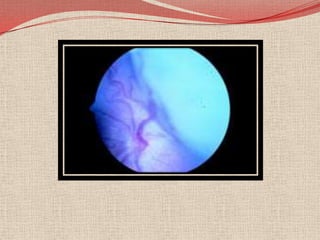


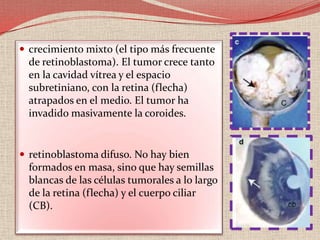
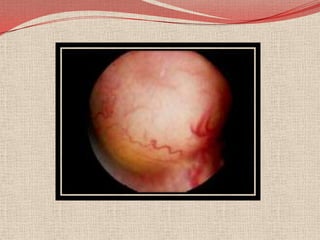
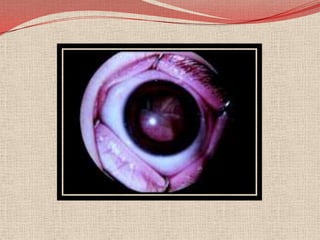
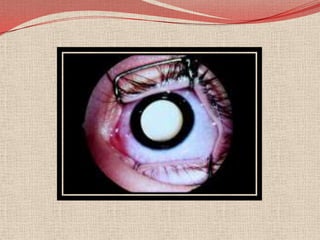
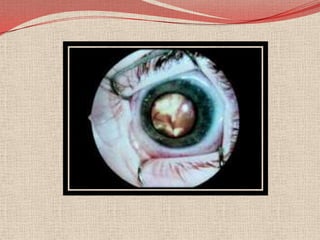

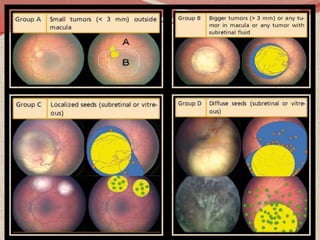








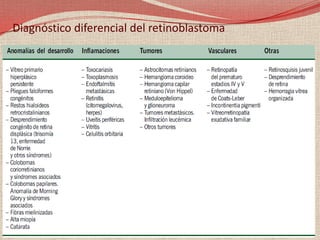
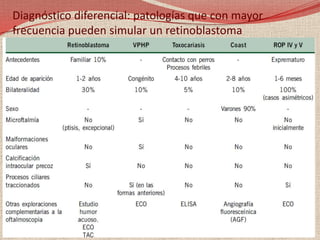










El retinoblastoma es un tipo de cáncer de la retina que principalmente afecta a niños pequeños y se asocia a mutaciones en el gen RB1. La detección temprana y los avances terapéuticos han mejorado la tasa de supervivencia al 91%, aunque la forma bilateral es más común en un porcentaje significativo de los casos. Los métodos de diagnóstico y tratamiento continúan evolucionando, buscando mejorar la calidad de vida y las perspectivas futuras de los pacientes.






















































![Ppt0000016 [SóLo Lectura]](https://cdn.slidesharecdn.com/ss_thumbnails/ppt0000016slolectura-100126154744-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)
































