Descargado 950 veces
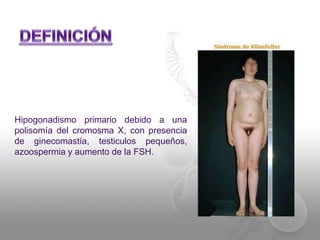



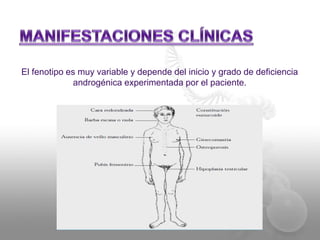
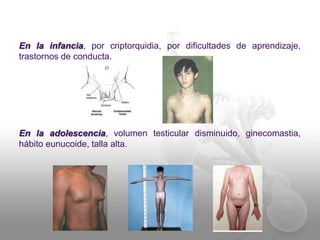
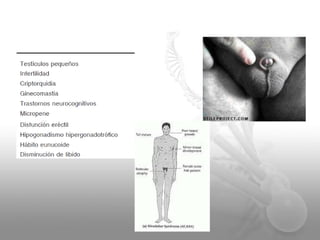
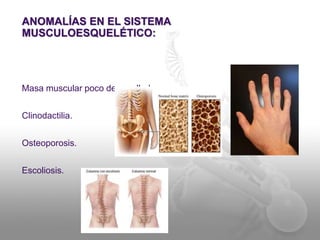

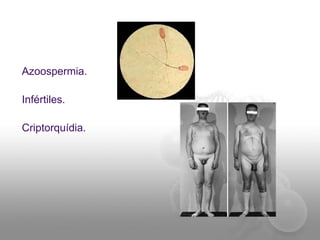















El documento describe el síndrome de Klinefelter (SK), una condición genética causada por la presencia de al menos un cromosoma X extra en los hombres. Los principales síntomas incluyen hipogonadismo, ginecomastia, testículos pequeños y azoospermia. El SK se asocia con un mayor riesgo de osteoporosis, cáncer de mama, enfermedades autoinmunes y problemas de desarrollo e intelectual. El tratamiento consiste en reemplazo de testosterona de por vida para prevenir complicaciones.



































































