Descargado 1595 veces







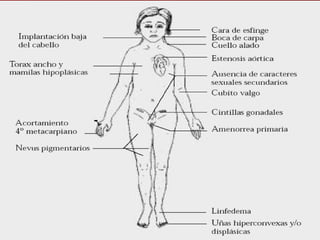


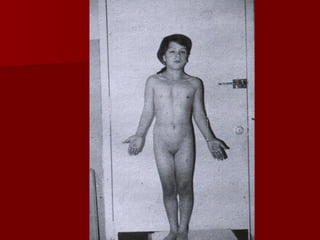
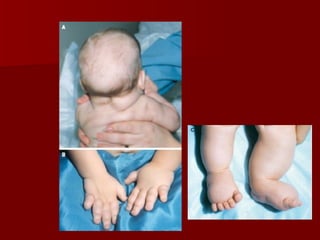

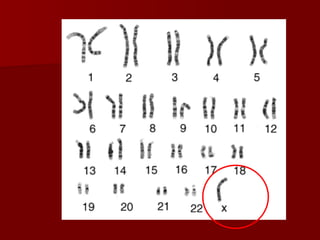






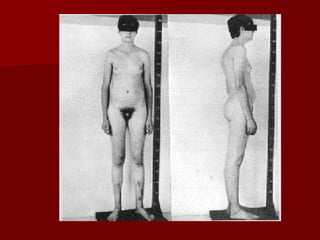











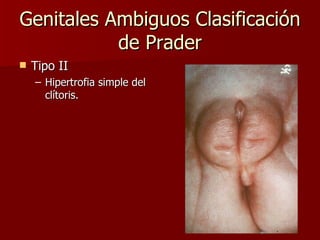

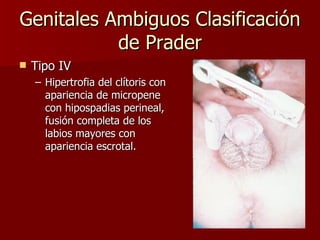




El documento describe varios síndromes relacionados con la diferenciación sexual anormal, incluyendo el síndrome de Turner, el síndrome de Klinefelter, genitales ambiguos y pseudohermafroditismo. Describe los hallazgos clínicos, causas, diagnóstico y tratamiento de cada condición.



































































































