Descargado 121 veces






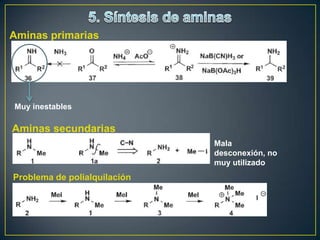
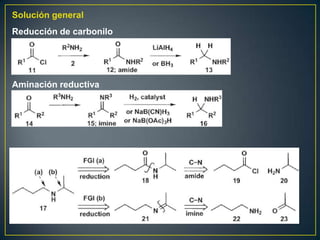
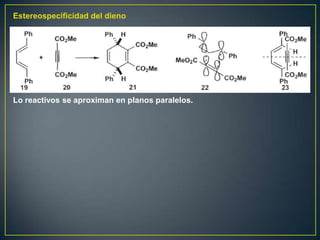
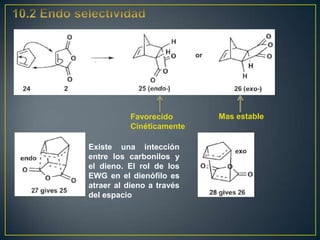
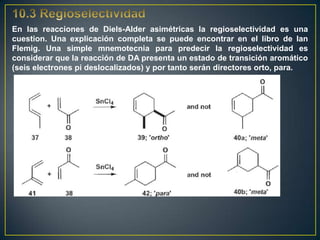
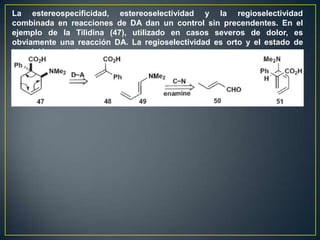

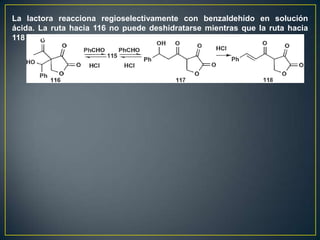

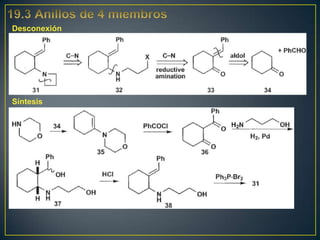
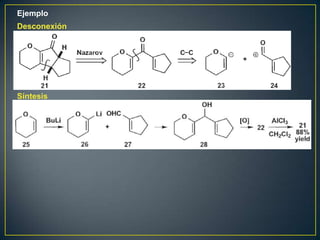
![Un rearreglo sigmatrópico es una reacción unimolecular en la cual un enlace
sigma se mueve de un lugar en una molécula a otra. En esta reacción no hay
cambio en el número de enlaces sigma. Los rearrelgos se pueden clasificar por
la numeración de los antiguos enlaces sigma y numerando en la dirección en
que se encuentra el nuevo enlace sigma.
Rearreglo
Vinil ciclopropano a ciclopenteno sigmatrópico [3,3]
Rearreglo [1,3]
La reacción procede con calentamiento fuerte >300°C. Existe una discrepancia
con respecto al mecanismo. Unas personas piensan que es concertada
mientras que otros dicen que es radicalario](https://image.slidesharecdn.com/sntesisorgnica-desonexiones-121024104143-phpapp01/85/Sintesis-organica-desonexiones-174-320.jpg)
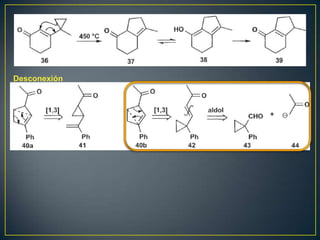
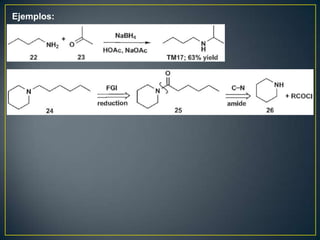
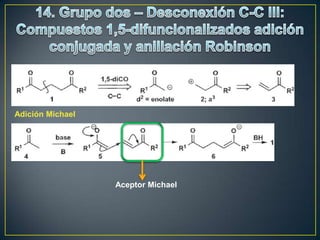
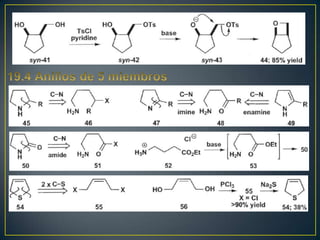
![Rearreglos sigmatrópicos-[3,3]
X= H, R, HO, OR, NH2 La reacción
funciona bien](https://image.slidesharecdn.com/sntesisorgnica-desonexiones-121024104143-phpapp01/85/Sintesis-organica-desonexiones-176-320.jpg)
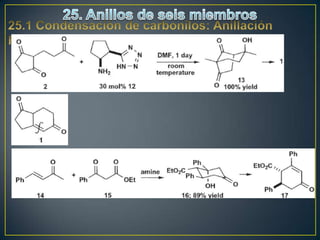
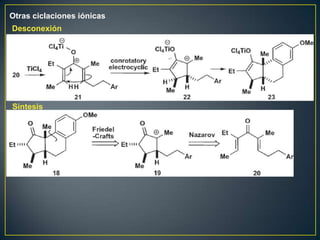
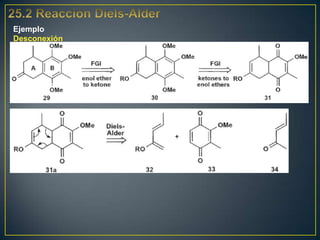
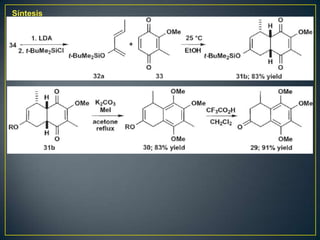
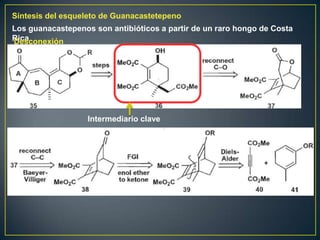
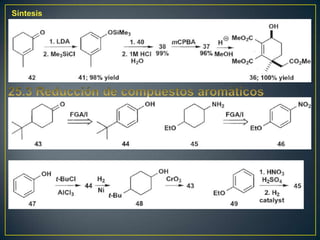
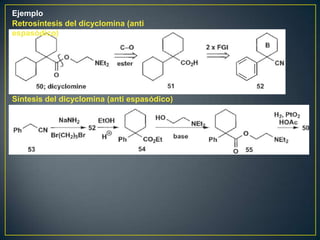
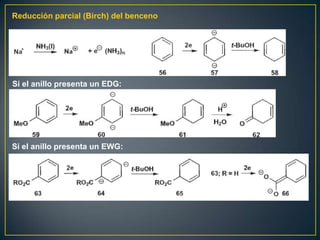
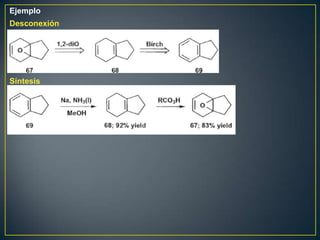
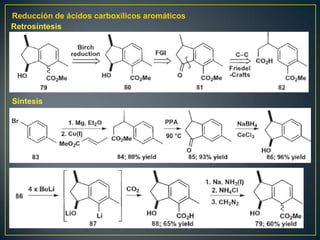


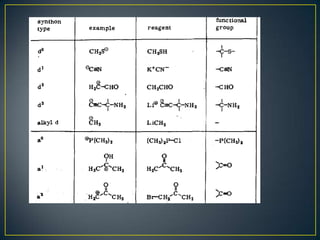
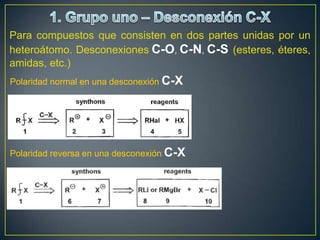
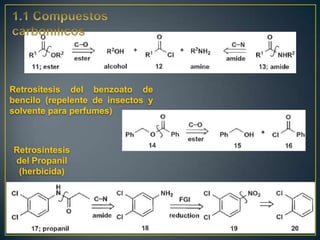
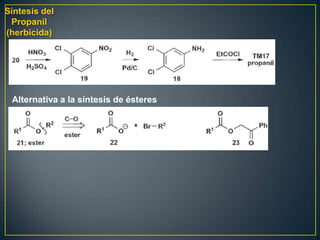
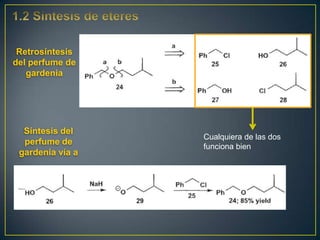
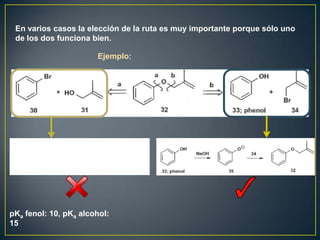
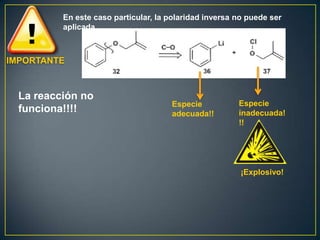
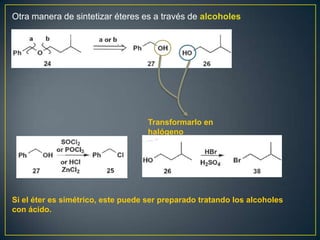
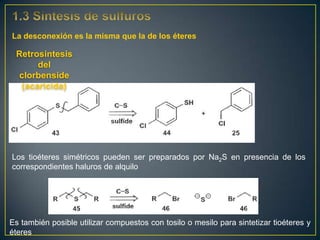
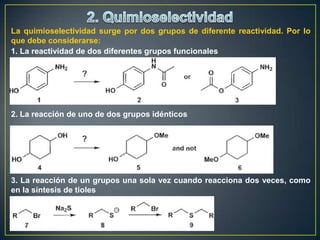
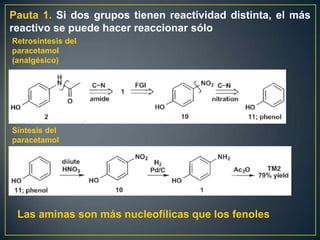
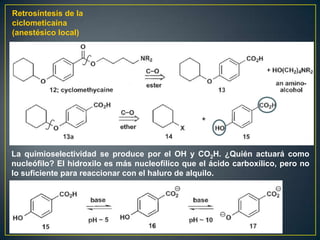
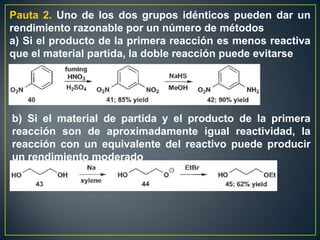
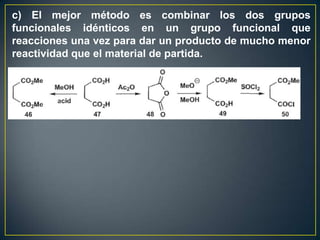
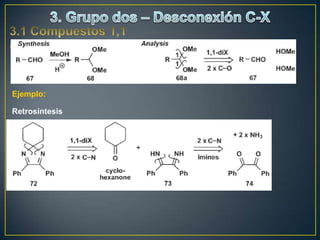
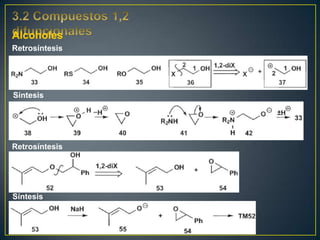
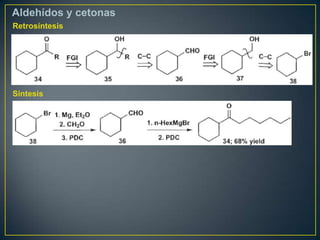
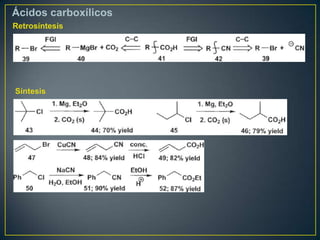
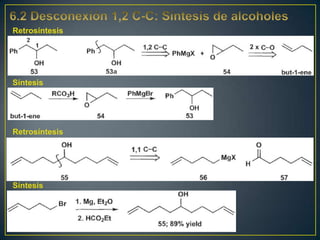
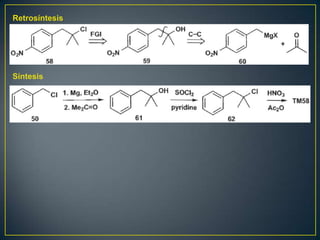
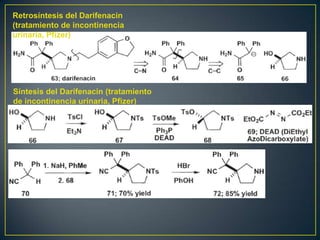
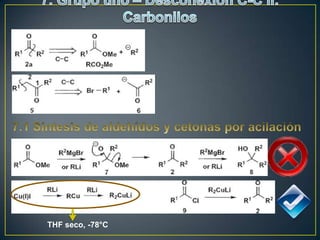
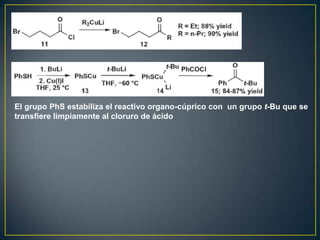
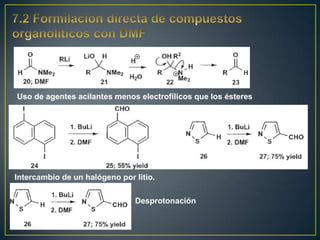
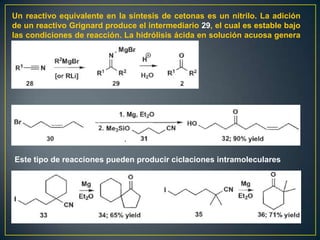
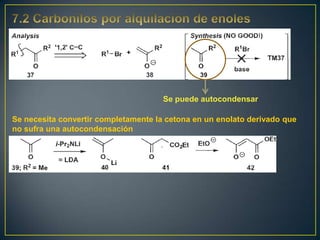
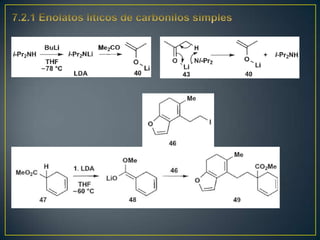
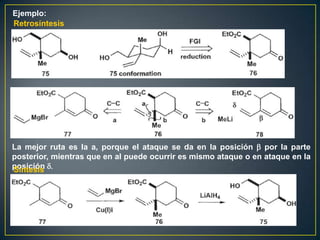
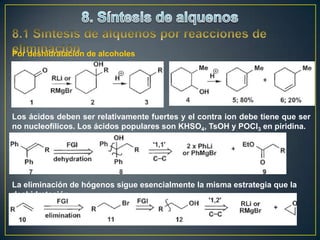
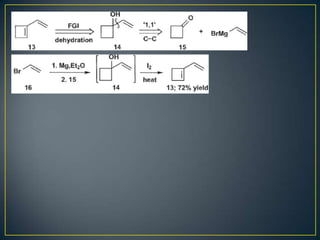
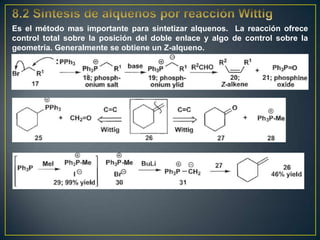
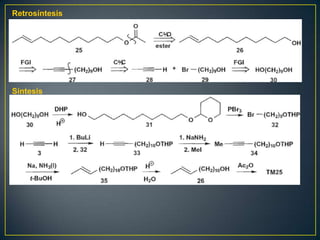
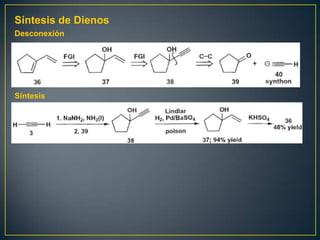
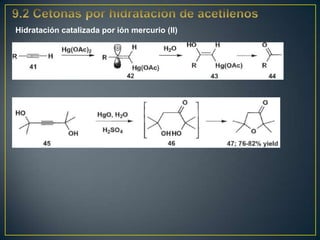
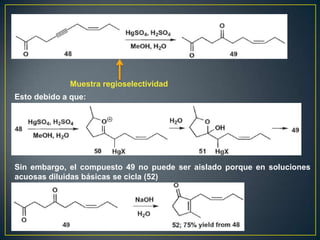
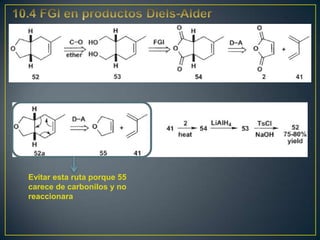
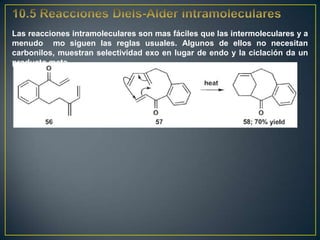
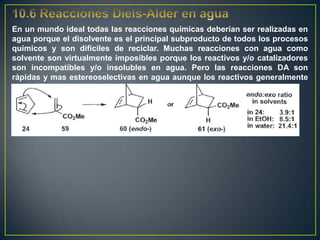
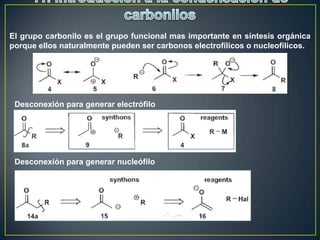
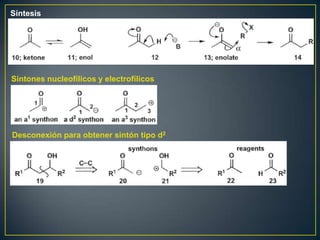
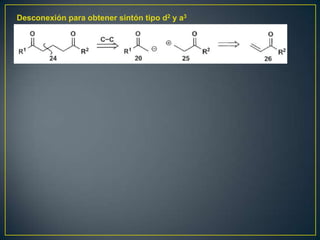
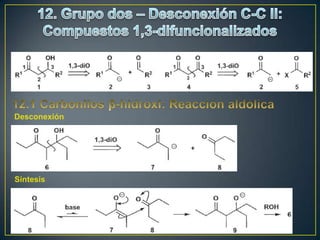
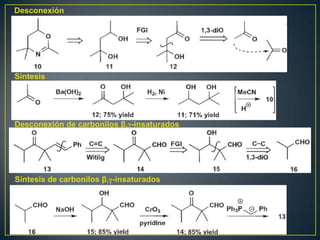
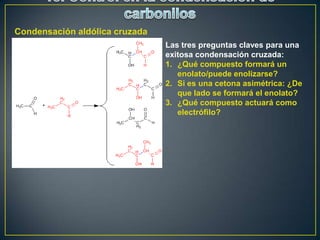
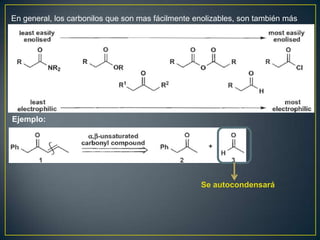
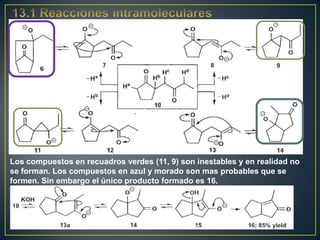
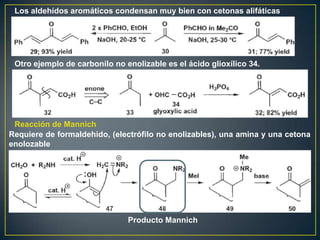
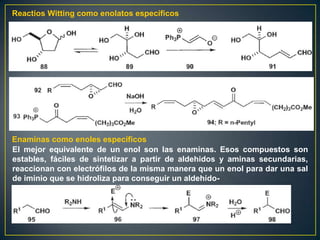
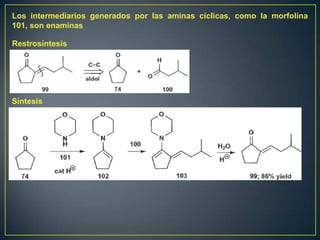
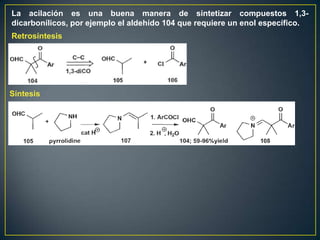
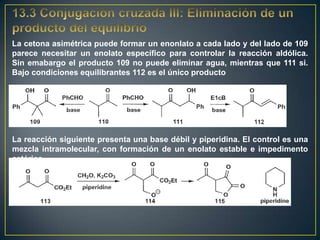
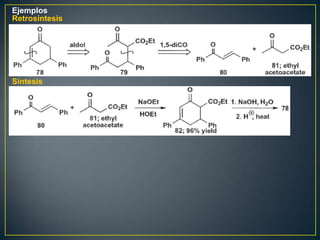
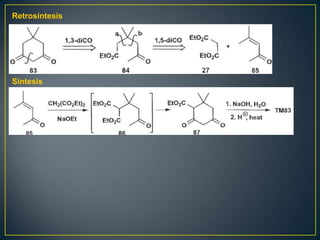
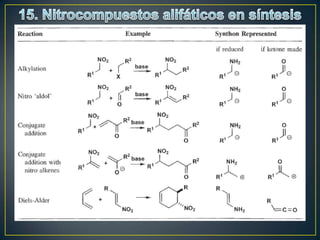
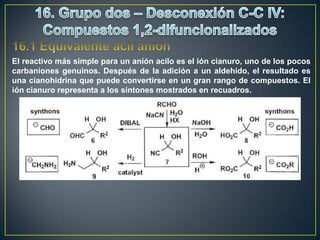
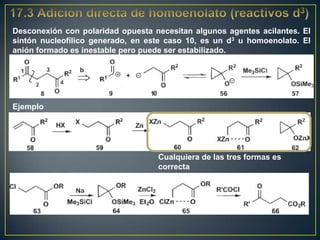
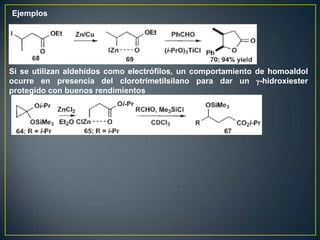
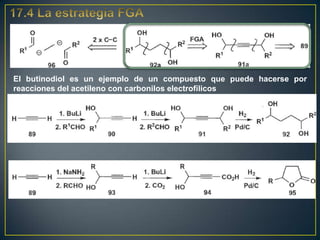
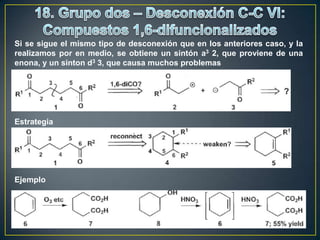
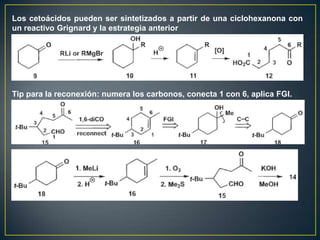
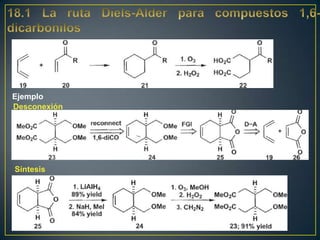
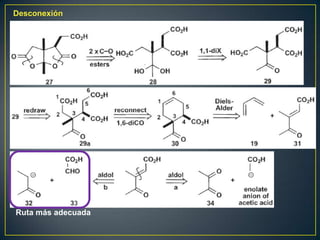
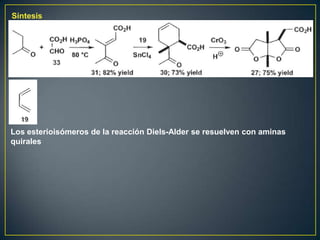
Este documento presenta una introducción a la síntesis orgánica y los conceptos de análisis retrosintético. Explica conceptos como flechas de retrosíntesis, desconexión de enlaces, polaridad normal y reversa, y el uso de sintones. Incluye ejemplos de retrosíntesis y síntesis de varios compuestos orgánicos como benzoato de bencilo, propanil y perfume de gardenia.























































































