
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Similar a Patologia Del Sistema Linfopoyetico
Similar a Patologia Del Sistema Linfopoyetico (20)
Último
Último (20)
Patologia Del Sistema Linfopoyetico
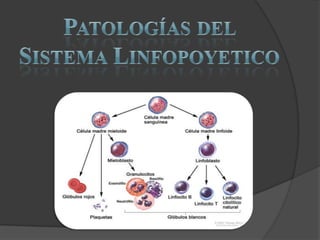
- 1. Patologías del Sistema Linfopoyetico
- 2. Leucopenia Disminucióndel número de leucocitostotalespordebajode 4.000 - 4.500 /mm³
- 3. Neutropeniao granulocitopenia< 1800/ μL Linfopenia< 1,000 / μL Eosionopenia< 50 /mm³; se ve en el Síndrome de Cushing y con el uso de ciertosmedicamentos (como los corticosteroides) Monocitopenia< 100 /mm³; Presente en anemia aplásica.
- 4. Neutropenia Masfrecuente de lasleucopenias. Valor normal: 2.000 y 7.500/mL Neutropenia < 1800/ μL Riesgo de Infección -Severa(< 500/μL) -Moderado(500 to 1000/μL) -Leve(1000 to 1500/μL)
- 5. Causas Produccióndisminuida o ineficaz Inhibición de celulasmadresprecursorasmielodeas y procesoinfiltrante de medulaosea. (se acompañan de anemia y trombocitopenia) Fármacosqueinhibencélulasprecursoras
- 6. Procesospatológicosporgranulopoyesisdefectuosa. (anemia megaloblasticapordeficiencia de vitamina B12 y folato) síndromesmielodisplásicos con destrucciónintramedular de precursores. RaravezcausahereditariasSíndrome de Kostmann (autosómicorecesivo, agranulocitosis)
- 7. Destrucción o disminuciónacelerada Mecanismosinmunitarios(LES y farmacos) Secuestroesplénico Usoexcesivoperiféricoporinfecciones graves
- 8. NeutropeniaporFármacos Causamásfrecuente Quimioterapia y Radioterapiaesprevisible y relacionada con la dosis. Aminopirina, cloranfenicol, sulfamidas, tiouracilo, clorpromazina y fenilbutazonaforma idiosincrática
- 9. Casos de destrucciónmasiva de neutrófilosmaduros y agranulopoyesisineficazcausahipercelularidad de médulaósea. Agranulocitosisporagentesqueinhibencrecimiento o destruyenprecursorespresentancifrasbajas de elementosleucopoyeticosqueestánmadurando.
- 10. RasgoCaracteristico Infecciones Lesionesnecrosantes y ulceras(suelo de la boca, encias y mucosa oral) Lugaresmenosfrecuentespiel, tubodigestivo, pulmon, riñones y vagina
- 12. Linfocitopenia Normal 1100 μL < 1,000 / μL sangre se encuentranlinfocitos B (25%) y células T (75%) y 65% son linfocitos T auxiliares (CD4). Asintomatica, puedeindicarcontagiopor VIH Riesgo de cancer
- 13. Causas Menorproducciónde linfocitos Mayor destrucciónde linfocitos Perdida de linfocitos (Enfermedad de Whippie)
- 14. Neumoníaporvaricela NeumoníaPneumocystisjiroveci Lesion de sarcoma de Kaposi
- 15. proliferaciónreactiva (inflamatoria) de leucocitos y ganglioslinfaticos Leucocitosis: Aumentotransitorio en el número de los leucocitos de la sangre (mayor de 10.000/mm3). Reacciónque se observa en diversosestadosinflamatorios
- 16. hemostacia de leucositosesmediadaporfactores de crecimiento, citocinas y moléculas de adhesión Inflamaciónaguda: (1-IL) y (TNF) liberanleucocitos de fondocomún Inflamacióncrónica: (CSF) y (TNF) aumentan el fondocomún de célulasprecursoras .
- 20. Linfadenitis Inflamación de uno o másganglioslinfáticos. Los gaglioslinfáticossufrencambiosreactivossiempreque son estimuladosporagentesmicrobiológicos, desechoscelulares o cuerposextrañosquepenetranunaherida o la circulación.
- 21. LinfadenitisInespecificaAguda Drenajedirecto de los microorganismo Ganglio cervical principal Gangliosaxilares e inguinales Gangliosmesentéricos
- 22. Morfologia Macroscopicamente: ganglioslinfaticoshinchadose ingurgitados y son de color gris-rojizo Microscopicamente: reciduos de bacterias o célulasnecroticas. Agentepiogenoconvierte el centro del foliculo en pus. Células de revestimiento de senos se hipertrofian, se vuelvencilíndricas y puedehaberhiperplasia.
- 23. Los ganglioslinfáticoaumentan de tamañodebido a la infiltracióncelular y al edema.
- 24. LinfadenitisInespesificaCrónica HiperplasiaFolicular -Activalinfocitos B -Centrosgerminales de numero y tamaño variable. -Numerosas mitosis y macrofagos.
- 25. Hiperplasiaparafolicular Difenilhidantoina Infección viral Vacuna anti-viral
- 26. Hiperplasia Reticular HistocitosisSinusal Distención y resalte de los sinusoideslinfaticos En carcinoma de mama estapresente, comorespuesta immune contra el tumor.
- 27. Enf. de arañazo de gato Toxoplasmosis
- 29. Hiperplasia ganglionar reactiva Proliferaciones benignas de las células de uno o más de los diferentes compartimientos anatómicos e inmunológicos de los tejidos linfoides.
- 30. Su causa es generalmente desconocida, pero muchas de ellas son provocadas por agentes bacterianos o virales o de sus productos. El aspecto morfológico de las hiperplasias pueden variar acorde a la edad, capacidad inmunológica y contacto previo con el agente. También puede variar por el tiempo de exposición y duración del estímulo.
- 31. Pueden comprometer: folículos (zonas B) Áreas interfoliculares o Paracorticales timo dependientes (células T) Células de los senos que pertenecen al sistema macrófago-monocitoide.
- 32. Los caracteres morfológicos comunes de las hiperplasias son: Conservación de la arquitectura fundamental del ganglio Expansión de alguna de las zonas o compartimientos del ganglio Proliferación policlonal de los elementos linfoides o de estirpe macrofágica.
- 33. Linfadenitis Inespecífica Aguda Esta clase de linfadenitis es causada generalmente por agentes bacterianos o virales, que penetraron en una herida o en la circulación Manifestaciones clínicas: Fiebre, dolor abdominal, malestar general. Macroscópicamente se observan ganglios hinchados e ingurgitados, color gris rojizo.
- 34. Microscópicamente se destacan los folículos linfoides, grandes centros germinales donde abundan mitosis. Histiocitos con partículas de bacterias en su interior. Puede presentar necrosis en folículos.
- 35. En reacciones menos graves se observa a veces un infiltrado de neutrófilos rodeando a los folículos, y puede haber muchos neutrófilos en los senos linfoides.
- 36. Linfadenitis crónica inespecífica Es la inflamación de los ganglios linfáticos cursado de manera crónica. Entre las manifestaciones clínicas generales se encuentran: Nódulos linfáticos inflamados dolorosos, sensibles o duros La piel sobre un nódulo rojiza caliente al tacto Fiebre con síntomas de escalofríos Pérdida del apetito Transpiración abundante Pulso rápido Debilidad general Dificultad para tragar Dificultad para respirar Rigidez en el cuello.
- 37. Hiperplasia Folicular Proceso inflamatorio que activa a las células B ante un estímulo, con un aumento de los ganglios regionales. Macroscópicamente se observa un ganglio aumentado de tamaño, generalmente entre 1,5 y 2,5 cm.; en casos excepcionales puede medir más de 4 cm. Se reconoce aumento en el número y tamaño de los folículos.
- 38. Microscópicamente Se observan centros germinales grandes, redondos o alargados (folículos secundarios) y de aspecto abultado. En los polos opuestos de un centro germinal reactivos se pueden distinguir dos regiones: Una zona oscura, que contiene células B proliferantes parecidas a blastos (centroblastos). Una zona clara, formada por células B con contornos nucleares hendidos o irregulares (centrocitos).
- 39. Hiperplasia Paracortical o Interfolicular Caracterizada por la ubicación de los cambios reactivos, los cuales se producen en los lugares del ganglio ocupados por las células T.
- 40. Histológicamente se observan: Células T que invaden y a veces parece que borran los folículos de las células B En regiones interfoliculares: células T activadas (inmunoblastos) dispersas, tres a cuatro veces más grandes que los linfocitos en reposo. Hipertrofia de las células endoteliales e los vasos y sinusoides Infiltrado celular mixto formado principalmente por macrófagos y a veces eosinófilos. Inmunohistoquímica: CD4+
- 41. Se presenta en las reacciones inmunitarias inducidas por fármacos, en las infecciones virales agudas, y especialmente en la mononucleosis infecciosa; en lupus eritematoso sistémico y después de las vacunaciones contra algunas enfermedades virales.
- 42. Hiperplasia Sinusal La Histiocitosis de los senos se refiere a la distensión y resalte de los sinusoides linfáticos. Es inespecífica, pero puede ser especialmente acusada en los ganglios de un cáncer.
- 43. Histológicamente se observa que las células del revestimiento endotelial están muy hipertrofiadas y los senos pueden estar prácticamente repletos de histiocitos.
- 44. Esta asociado a procesos neoplásicos como cáncer de mama. También puede presentarse en anemias hemolíticas y post transfusión.
- 45. Hiperplasia mixta Hay hiperplasia folicular e interfolicular; a veces se agrega la hiperplasia sinusal. Ejemplos: mononucleosis infecciosa, toxoplasmosis.
- 46. LINFOMAS
- 50. Los linfomas (ML) grupo heterogeneo de neoplasias malignas caracterizadas por proliferaciones derivadas de celulas nativas del sistema linfoide (los linfocitos -B y T, y celulas derivados de estos- e histiocitos y celulas NK), que por su localizacion pueden clasificarse como nodales (o ganglionares: GL y tejido linfatico) o extranodales (o extraganglionares: timo, el bazo, tejido linfoide asociado a la mucosa (MALT): tubo digestivo (GALT), pulmón (BALT), piel bazo, médula hematopoyética, dondequiera que se encuentre tejido linfoide asociado a las mucosas (MALT). LINFOMAS
- 51. Generalmente formando una masa tumoral, que luego puede extenderse al bazo, hígado, médula ósea y otros órganos en forma de tumores metastásicos. Rara vez, el linfoma término se utiliza para referirse a una proliferación maligna de las células histiocítica. LINFOMA ↔ LEUCEMIA LINFOMAS
- 53. El linfoma de Hodgkin –LH (enfermedad de Hodgkin)
- 54. El linfoma no-Hodgkin –LNHRepresenta alrededor de 35 tipos de cáncer que afectan a las células del sistema inmunológico. Hay 5 subtipos de la enfermedad de Hodgkin y alrededor de 30 subtipos de linfoma no Hodgkin
- 55. DIFERENCIAS CLINICAS ENTRE LA ENFERMEDAD DE HODGKIN Y LOS LINFOMAS NO HODGKINIANOS
- 57. LINFOMAS Se puede sospechar las neoplasias linfoides por características clínicas pero el diagnostico requiere examen histológico de GL y Tejidos afectados En la mayoría de Neoplasias linfoides, las células hijas derivadas del progenitor maligno comparten la misma configuración y la misma secuencia de genes de receptores de antígenos y sintetizan idénticas proteínas receptores de antígenos (Ig o R.C.T.) La mayoría de neoplasias linfoides (80-85%) son de origen B, y la mayor parte de las restantes son tumores de células T, y son raras los tumores de células NK. Frecuente alteración de la arquitectura normal y función del sistema inmune, con anomalías inmunológicas. Las células B y T neoplasias tienden a recapitular el comportamiento de equivalente normales. (localización y regulación ganglionar, diseminación)
- 58. LINFOMAS EPIDEMIOLOGIA Tumor maligno más frecuente en los Estados Unidos, y el tercero más común en los niños. El linfoma puede ocurrir a cualquier edad, incluso en la infancia. El linfoma es más frecuente en varones que en mujeres (1.5:1) para el LH y LNH Cerca de 24.000 personas mueren de linfoma no Hodgkin y HL 1400 de cada año. EL NationalCancerInstitute (NCI) estima 8,510 casos nuevos y 1,290 muertes LH y 65, 980 casos nuevos y 19,500 muertes por LNH en el año 2009
- 60. LINFOMAS EPIDEMIOLOGIA El linfoma no Hodgkin es mucho más común que la enfermedad de Hodgkin, sin embargo el LH presenta mayores casos avanzados. La incidencia del linfoma no Hodgkin aumenta progresivamente con la edad (pico de aproximadamente 60 años). La enfermedad de Hodgkin es más común en personas entre 16-34 años (adultos jovenes) y en personas con ≥ 55 años.
- 61. Etiologia. Factores etiológicos Edad. Generalmente el riesgo de LNH aumenta con la edad. El LH esta asociado a un mal pronostico en los ancianos comparado con pacientes mas jóvenes. Infecciones por Virus Epstein Barr (VEB)-L. Burkitt-, VIH, Virus linfocitico-T humano tipo 1 (HTLV-1), Helicobacterpylori-MALTomagastrico- y Virus de la Hepatitis B o C, Mycobacterium tuberculosis Inmunosupresión (VIH, enfermedad inmune, terapia inmunosupresora post transplante, heredadatalengectasia, ataxia, síndrome de Klinefelter, síndrome de Chédiak-Higashi , síndrome de Wiskott-Aldrich; desordenes autoinmunes (reumatoide, enfermedad celiaca, síndrome de Sjögren, LES) Exposición aproductos tóxicos. Ocupacionales (plaguicidas, herbicidas, tintes para cabello, difenilhidantoina- hiperplasia linfoide-linfoma-) industriales (arsénico, clorofenoles, solventes orgánicos, plomo, cloruro de vinilo y asbestos.) Genéticos (Antecedentes familiares de enfermedad de Hodgkin o LNH) Radioterapia o la quimioterapia
- 63. ESTADIFICACION. Ann Harbor Estadio I (enfermedad temprana) linfoma situado en una sola región de ganglios o un órgano extranodal. Estadio II (enfermedad localmente avanzada). El linfoma se encuentra ubicado en dos o mas regiones de GL, ubicados en el mismo lado del diafragma o en una región de los GL y en órganos o tejidos cercanos. Estadio III (enfermedad avanzada). Linfoma afectando dos o mas regiones de GL o en un GL y un órgano a ambos lados del diafragma. Estadio IV. (enfermedad diseminada). Linfoma fuera de los GL y bazo con extensión a la hueso, médula ósea, o SNC
- 64. Estadío I: ganglios localizados Estadío II: más ganglios, supra- diafragmáticos Estadío III: más ganglios, en cualquier lugar Estadío IV: afectación extraganglionar MESESoAÑOS
- 65. CLASIFICACION SEGÚN GRADO. Bajo grado. son linfomas indolentes que crecen lentamente. Generalmente no requieren tratamiento inmediato, a menos que exista compromiso en la función de algún órgano. Rara vez se curan y pueden tranformarse con el tiempo a combinación de tipos de indolente y agresivo. Grado intermedio. Linfomas de crecimiento rápido con un patrón agresivo, que requieren tratamiento inmediato, y con frecuencia son curables. Alto grado. Presentan mayor rapidez de crecimiento que los de grado intermedio, que requieren inmediato e intensivo tratamiento, y son muy a menudo incurables
- 66. Hospital Guy. Londres, Inglaterra 1832 ENFERMEDAD DE HODGKIN
- 68. Mediastino anterior> superior> medio
- 70. La posibilidad de supervivencia de 5 años en pacientes con linfoma de Hodgkin en estadio I y II es del 90%, , III, 84%, y IV, 65%..
- 72. adultos jóvenes (15-34 años y) y las personas mayores (> 55 años).
- 74. El componente reactivo (linfocitos pequeños maduros [B: alrededor de folículos, T: alrededor de ceulas RS], eosinófilos, plasmocitos, histiocitos, células foliculares dendríticas, neutrófilos, fibroblastos, fibras colagenas, fibrosis y capilares).
- 75. HISTOLOGIA De anormalidades de linfocitos del linaje B, generalmente del centro germinal o post centro germinal. Tambien pueden surgir de celulas T (1.2%) células Reed-Sternberg (RS).Componen sólo el 1-50% de la masa total de células tumorales. Inducen la acumulacion de L.reactivos, histiocitos (macrofagos), y granulocitos
- 77. Los tipos de células RS son: Clásica: Se observa en variantes Celularidad mixta, en la variante con disminución de linfocitos, y en la esclerosis nodular, principalmente.
- 78. Variante LP(Linfohistocitario)o L&H (“pop corn”): núcleos únicos o multilobulados, con nucléolos pequeños puntiformes, cromatina delgada, citoplasma escaso y nodular. Se encuentra en la variante de predominio linfocitico.
- 79. Variante pleomorfica. Núcleo único, multinucleado o multilobulado; nucléolos variables y prominentes; paracromatina variable y clara o hipercromatica, citoplasma moderado o abundante y eosinofilo. Se ve en la variante de depleción linfocitica
- 80. Mononuclear: Núcleo único, con nucléolos grandes (>1/3 de diámetro del núcleo) eosinofilos, paracromatina abundante y clara, citoplasma entre rosa y pálido. Puede observarse en cualquier variante.
- 81. Célula lacunar: núcleo único o multilobulado, nucléolo variable (generalmente <1/3 de diámetro del núcleo), eosinofilos., con paracromatina delgada o clara, con citoplasma abundante y pálido. En la esclerosis nodular, y pueden verse parecidas, en ocasiones, en la variante Celularidad mixta
- 82. CLASIFICACION la Organización Mundial de la Salud (OMS) 2001 No clásicas Predominante linfocitico (~5% casos) Favorable Clásico (~95%) Esclerosis nodular (60 - 80% casos) Favorable Linfocitos-rica (5% casos) Favorable Celularidad mixta (15 - 30% casos) Favorable Deplecionlinfocitica(<1% casos) desfavorable
- 83. CLASIFICACION DE LA ENFERMEDAD DE HODGKIN
- 85. Clásico. Esclerosis nodular. Mujeres (80%) afectacion mediastino, Estadio II Diseminnaciona bazo, hígado, MO y GL cervicales inferiores, supraclaviculares y mediastínico. HISTOLOGIA. bandas de fibrosis esclerosantes con colágeno (patron nodular) la capsula esta engrosada. Presenta la variante RS lacunarmás que la RS clásica con un fondo polimorfo de linfocitos T pequeños, eosinofilos, células plasmáticas y macrófagos. INMUNOFENOTIPO. CD15,30+ y CD45-,
- 87. Clásico. Celularidad mixta. Hombres (70%) de edad mayor con presentación en fase tardia (III y IV) con síntomas B sistémicos Ganglios linfáticos del abdomen y el bazo HISTOLOGIA. Infiltrado celular heterogéneo inflamatorio formado por linfocitos, eosinofilos, células plasmáticas y macrófagos benignos entremezclados con numerosas células RS y de la variedad mononuclear. Cambia la estructura normal. INMUNOFENOTIPO. CD15,30+ y CD45- Con pronostico favorable Asociado a VIH
- 89. Clásico. Rico de predominio linfocitico Hombres Clínicamente, la presentación y los patrones de supervivencia son similares a aquellos para la variante celularidad mixta. HISTOLOGIA. Se caracteriza por las células clásicas de Hodgkin y RS con un abundante trasfondo de linfocitos pequeños. Faltas células inflamatorias y bandas de colageno
- 91. Clásico. Deplecionlinfocitica Asociada a VIH, y a la edad avanzada Virus de Epstein Barr (VEB) se expresan en muchos de estos tumores Organosabdominales, GL retroperitoneales y la medula osea Menos comunes adenopatías periféricas 70% fase avanzada y 80%, síntomas B. HISTOLOGIA. Infiltrado difuso con pocos linfocitos o fibrosis. dos subtipos: sarcomatoso con numerosas células RS bizarras; y una variante fibrosis difusa con fibrosis extensa desordenada y raras RS, linfocitos raros, y con frecuencia pocos y fácilmente identificables células RS en un fondo hipocelular. Linfocitos prácticamente inexistentes
- 93. No clásica. Predominante linfocitico Pacientes <35 años adenopatías cervicales o axilares pocas veces afectación mediastinica y rara vez medular. HISTOLOGIA. . Celulas L&H, fondo predominantemente con linfocitos benignos con escasos eosinofilos, neutrofilos y células plasmáticas y con pocos signos de necrosis o fibrosis INMUNOFENOTIPO. Células tumoralesCD45,20 + y CD30-. 3-5% evolucionen linfomas difusos de células grandes Pronostico bueno.
- 96. CLASIFICACION HISTOPATOLOGICA OMS B. FORMAS CLASICAS (CD15,30+,20- A. PREDOMINIO LINFOCITICO NODULAR Edad: 43 años. Enfermedad localizada. Sobrevidas > 95%. VEB no se relaciona CD 15 -, CD 45 +. RICA EN LINFOCITOS (3%) Niños 10% a 15%. Adultos 7%. Comúnmente enfermedad localizada. VEB 10% CD15 - ESCLEROSIS NODULAR (67%) Jóvenes 40%. Adolescentes 70% Adultos 21%. Comúnmente ganglios cervicales y mediastinales. DEPLECIÓN LINFOIDE (Raros) Raro en niños. Adultos 11%. Enfermedad diseminada y afección a MO. VEB Raro CELULARIDAD MIXTA (27%) Niños 30%. Adultos 61%. Frecuentemente enfermedad avanzada con extensión extranodal. VEB 34% CD15 + VEB 96% CD15 +
- 97. MANIFESTACIONES CLINICAS Linfadenopatía asintomática (80%), Este agrandamiento de los ganglios linfático puede causar secundariamente síntomas por compresión de vena, nervio, o el estomago. La ictericia intrahepática, Disnea, tos y sibilancias por compresión traqueobronquial, también la esplenomegalia y hepatomegalia. Dolor por alcohol. " B "( pérdida de peso inexplicable [> 10% del peso corporal en los últimos 6 meses] y apetito, en un 10% del peso total , fiebre, sudores nocturnos fiebre de Pel-Epstein sindromesparaneoplasicos, incluyendo la degeneración cerebelosa, neuropatía, síndrome de Guillain-Barré, o leucoencefalopatía multifocal.
- 98. DIAGNOSTICO Radiografía de tórax TC de tórax, abdomen y pelvis Hemograma, VSG, fosfatasa alcalina, LDH, pruebas de función hepática, albúmina, calcio, urea y creatinina Biopsia del ganglio linfático Biopsia de médula ósea Posiblemente PET para la estadificación, la gammagrafía ósea si están presentes los síntomas del dolor óseo, o resonancia magnética si neurológicos
- 99. Tratamiento Estadio I Estadio II Estadio III 90% 85% Radioterapia Quimioterapia Estadio IV III-A III-B 70-80% 75-80% Quimioterapia Radioterapia Quimioterapia 50% MOPP ABVD
- 100. PRONOSTICO El sexo masculino Edad de ≥ 45 años La enfermedad en estadio IV La albúmina (examen de sangre) a < 4,0 g / dL La hemoglobina a < 10,5 g / dL Globulos blancos (leucocitos) > 15.000 / ml o >15x 109/L Bajo recuento de linfocitos (inferior a 600/mLo< 0.6x109/L ; < 8% del recuento total de leucocitos) EVALUACIÓN: 2 a 3 factores: BUENO; 4 factores: MALO, >5 factores : PÉSIMO
- 102. LINFOMAS DE LOS LINFOCITOS B Y T (LINFOMAS NO HODGKINIANOS)
- 103. LINFOMAS NO HODGKINIANOS Grupo heterogéneo neoplasias malignas clonaleslinfoproliferativas del tejido linfoide extramedularcon diferentes patrones de comportamiento y de respuesta al tratamiento Diseminacion a sitios adyacentes y a distancia por la circulación linfática y finalmente por vía sanguínea. La mayoría (85%) resulta de una proliferación de células B, sólo el 15% se deriva de T / NK. 5 veces más frecuente que la EH. Pueden ser indolentes o agresivos.
- 104. Comprende todos los linfomas cuya histología no muestra la célula de Reed-Sternberg y es muy heterogéneo. No todos se localizan en ganglios linfáticos. Un ej. de tejidos extraganglionaresgeneradores de linfoma son los tejidos linfoides asociados a las mucosas.
- 106. Epidemiología 5 veces más frecuente que la enfermedad de Hodgkin. Predomina en varones de edad media: 20 a 40 años. Tipo histológico más frecuente, es el difuso de células B grandes, seguido del folicular. 50% de sobrevida a 10 años.
- 107. CLASIFICACION REAL I. Neoplasias de precursores de células B Leucemia / linfoma linfoblástico de precursores B II. Neoplasias de precursores de células T Leucemia / linfoma linfoblástico de precursores T II. Neoplasias de células B periféricas (maduros) Leucemia linfocítica crónica / linfoma linfocítico de células pequeñas. Prolinfocitica de linfocitos B Linfoma linfoplasmocitario Linfoma de células del manto Tricoleucemia Mieloma de celulas plasmáticas Gammapatia monoclonal de significado incierto (GMSI) Linfoma folicular De grado citológico I De grado citológico II De grado citológico III Linfoma de la zona marginal Leucemia de células peludas Plasmocitoma solitario del Hueso Plasmocitomaextraoseo Amiloidosis primaria Enfermedad de cadenas pesadas Linfoma difuso de células B grandes Linfoma de linfocitos B de la zona marginal extraganglionar del tejido linfático asociado a la mucosa (Linfoma MALT o MALToma) Linfoma de linfocitos B de la zona marginal ganglionar Linfoma mediastinico (Timico) de linfocitos B grandes Linfoma intravascular de linfocitos B grandes. Linfoma primario de los derrames Linfoma/leucemia de Burkitt IV. Neoplasias de células T periféricas y NK Leucemia linfocítica crónica de células T Leucemia/ diseminada Leucemia prolinfocitica de células T Leucemia prolinfocitica de células T grandes y granulosas Leucemia agresiva de células NK Leucemia/ linfoma de células T del adulto Cutaneos Micosis fungoide Sindrome de Sézary Linfoma anaplasico cutáneo primario Linfoma de células grandes Papulosislinfomatoide Otros linfomas ganglionares Linfoma extraganglionar de células NK/T de tipo nasal Linfoma de células T del tipo enteropatico Linfoma de células T hepatoesplenico Linfoma de células T similar a la paniculitis subcutánea Ganglionares Linfoma angioinmunoblastico de células T Linfoma periférico de células T sin especificar Linfoma anaplasico de células grandes Neoplasias de estirpe y etapa de diferenciación inciertos Linfoma de células NKblasticas.
- 110. LINFOMA LINFOBLASTICO AGUDO Neoplasias maligna de células B o T inmaduras precursoras (linfoblastospreB y preT) en los ganglios. OMS: Leucemia linfoblástica de precursores B (LAL-B)/linfoma linfoblástico (LLB-B) Leucemia linfoblástica de precursores T (LAL-T)/linfoma linfoblástico (LLB-T)
- 111. EPIDEMIOLOGIA EEUU. Aprox. 2500 nuevos casos de LLA por años, la mayoria en niños de 15 años de edad. Incidencia de LLA-preB, alta hacia los 4 años, con un pico en la niñez temprana. Y LLA-preT en la adolescencia. El LLA es frecuente en los niños (30-50%) menores de 6 años (alrededor del 75% de los casos); en los adultos, el linfoma linfoblástico representa menos del 5% del total de linfomas no Hodgkin en adultos.
- 112. Predomina en hombres Linfoma linfoblástico, una masa tumoral compuesta de linfocitos precursores proviene de linfocitos T en el 90% de los casos. Forma típica de presentación: distress respiratorio en adolescentes. Restringido a región supradiafragmática con compromiso de GL axilares, supraclaviculares, cervicales pero sin afectar sangre periférica o Medula ósea.
- 113. De cels T inmaduras; algunos son de cels pre T o NK o incluso lineaje B Casi todas las neoplasias malignas de los precursores de linfocitos B son fundamentalmente leucémicas; el linfoma linfoblástico es infrecuente. HISTOLOGIA. Macro: tumor sólido, blando, no encapsulado, con preservación de estructura de timo; Micro: infiltra parénquima de timo y puede confundirse con timoma tipo B1
- 115. LINFOMAS DE LINFOCITOS B MADUROS PERIFERICO
- 116. Leucemia linfoblastica crónica (LLC) y linfoma linfocitico de células pequeñas (LLP) indistinguibles en morfología, genotipo, y fenotipo, pero se diferencian en el grado de linfocitosis que producen en la sangre periférica EPIDEMIOLOGIA LLC es el linfoma mas común en adultos del mundo occidental, y la LLP constituye el 4% de LNH. La mayoria de pacientes >50 años (promedio: 60 años) con un predominio en varones (2:1)
- 117. HISTOLOGIA. Sangre periferica. (LLC). células borrosas. Pueden afectar al bazo y el hígado. Ganglio: linfocitos predominantemente pequeños (6-12µ), nucleos redondeados o irregulares, con cromatina densa y citoplasma claro; y de prolinfocitos Estos últimos pueden formar aglomerados llamados centro de proliferación, de gran actividad mitótica. Estos centros son patonogmonicos de LLC y LLP.
- 118. MANIFESTACIONES CLINICAS. Asintomaticos. Frecuentes en LLC y LLP Sintomaticos. Inespecificos: cansancio fácil, perdida de peso y anorexia. Adenopatia generalizada y hepatoesplenomegalia (50-60% de casos) Cifra total de leucocitos elevada o llegar a 200,000/mm3 Hipogammaglobulinemia, con mayor suceptibilidad a infecciones bacterianas 15-20% pacientes con Ac antihematies (anemia hemolítica inmunitaria) o antiplaquetarios (trombocitopenia inmunitaria)Ac Igpoliclonales por células B autorreactivas no neoplasicas.
- 119. INMUNOFENOTIPO: CD19, CD20 +(Selectivos B), CD5 (de linfocitos T, expresado por un pequeño grupo de LB), expresión IgM (Cadenas pesadas Ig), y cadenas ligeras kappa o lambda. GENETICA. Las alteraciones cromosómicas se observan en un 20-30% de los casos. Son frecuentes la trisomia 12, deleciones de 13q 12-14, y delecion de 11q
- 120. PRONOSTICO. Variable, dependiente del estado clínico. Superviviencia media 4-6 años (pctes con masa tumoral minima ≥ 10 años) Trisomia 12 y delecion 11q, mal pronostico Tendencia agresiva, (supervivencia < 1 año): transformación prolinfocitica (15-30 % casos), o linfoma difuso de células B grandes (Sindrome de Richter, 10% casos)
- 121. Linfoma Folicular EPIDEMIOLOGIA Forma mas frecuente de LNH en los EE.UU Frecuente en la mediana edad Genero H = M Menos frecuente en Europa y Rara en países asiáticos
- 122. HISTOLOGIA Semejantes a linfocitos B normales de centros germinales. GL, patrón de crecimiento con predominio nodular o nodular y difuso. Se presentan dos tipos de células: Centrocitos. Celulas pequeña, Contornos nucleares irregulares o hendidos, citoplasma escaso. Prodominio. Centroblastos. Células mas grandes con cromatina nuclear laxa, varios nucléolos,
- 124. INMUNOFENOTIPO CD19,20,10 (CALLA) +, expresa Igmonotipicas de superficie. A diferencia del LLC, LLP, y linfoma de células del manto no expresan CD5. Ademas son BCL2 + (a diferencia de los linfocitos B de los centros foliculares que son BCL2 negativos) GENETICA Lo mas característico del Linfoma folicular es la traslocacion (14-18), que produce yuxtaposición del locus IgH del cromosoma 14 y del locus BCL2 del cromosoma 18, con sobreexpresión de BCL2(favorece la supervivencia del linfoma folicular )A un asi no es observado en todos los casos. Foliculos reactivos con células B apoptosicas y folículos neoplasicos sin células apoptosicas
- 125. MANIFESTACIONES CLINICAS Linfocitosis < 20,000/mm3 (10% casos) Afectacion medular (85% casos) , como conglomerados linfoides paratrabeculares. Afectacion esplénica (pulpa blanca) y hepática (espacios porta) Adenopatias indoloras que suelen ser generalizadas. Rara afectación extraganglionar (TGI, SNC, testiculo) Incurable con posibilidad de evolución insidiosa y fluctuante
- 126. PRONOSTICO y EVOLUCION Supervivencia media de 7-9 años, sin mejora con tratamientos enérgicos Superviviencia en trasnformacion maligna < 1 año 30-50% Casos con transformación histológica maligna, con transformación en la mayoria de los casos en linfoma difuso de células B Rara tranformacion agresiva parecida a leucemia o linfoma linfoblástico (traslocacion cromosómica del locus c-myc)
- 127. LINFOMA DIFUSO DE CELULAS B GRANDES Grupo heterogéneo de tumores que en conjunto constituyen el 20% de todos los LNH y el 60-70% de neoplasias linfoides agresivas. Leve predominio en hombres Distribucion amplia en edades: Aparece en promedio a los 60 años, pero también constituye el 5% de los linfomas de la infancia.
- 128. HISTOLOGIA Células relativamente grandes (4-5 vcs el diamentro de un linfocito pequeño,) con un patrón de crecimiento difuso, nucleoredondo u oval, vesiculoso por la cromatina limitada a la membrana nuclear. (multilobulados), 2-3 nucleolos centrales, citoplasma moderado palido o basofilo. En tumores mas anaplasicos se encuentran células multinucleadas con con nucléolos grandes, con aspecto de inclusión (similar a RS)
- 131. VARIANTES ESPECIALES Linfoma de células B grandes asociado a inmunodeficiencia. En inmunodeficiencia grave de linfocitos T. Las células B neoplasicas suelen ser Virus de Epstein Barr (VEB) positivos latente. Linfoma de células B grandes de las cavidades corporales. Se manifiestan por derrames pleurales o ascíticos malignos.VIH, y ancianos En todos los casos las células están infectadas por el virus herpético humano 8.(asociado también en la etiología del sarcoma de Kaposi.)
- 132. INMUNOFENOTIPO CD19, 20, CD10 (N. originadas en células centrofoliculares), la mayoria tiene Ig de superficie, y Todos son TdT GENETICA El 30% la t(14;18), (característica del linfoma folicular) 20-30%, presenta traslocacion que fracturan el locus BCL6 del cromosoma 3..
- 133. MANIFESTACIONES CLINICAS Masa de crecimiento rápido Primera manifestación en TGI, piel, hueso y cerebro, y también pueden estar infectados, el anillo de Waldeyer y tejidos linfáticos de la orofaringe: amígdalas y adenoides. Puede haber masas destructivas en bazo e hígado Afectaciontardia de la medula osea, raro cuadro leucémico.
- 134. PRONOSTICO Los linfomas difusos de células B grandes producen rápida muerte sin tratamiento Con quimioterapia combinada intensiva se alcanza una remisión en el 60-80% de los pacientes y alrededor del 50% permanecen asintomáticos durante varios años, pudiéndose considerar curados Masa circunscrita, buen pronostico (diseminadas o voluminosas, mal pronostico) Tumores con reordenamiento del BCL6, buen pronostico Mutaciones en el P53, mal pronostico
- 135. Linfoma de Burkitt Linfoma linfoblástico, observado principalmente en niños africanos. Niños y adultos. 30% de LNH en niños en EE.UU. Asociado a infección por VEB. Muestra una translocación cromosómica 14q+/8q- (locus inmunogénico, y oncogenmyc). Tumor mandibular. Afectación GI. Histología: aspecto de “cielo estrellado”. Remisiones espectaculares con una sola dosis de citotóxico, como ciclofosfamida, 30 mg/kg IV.
- 138. CLASIFICACION Presentan similaridad histológicas, pero diferencias clínicas, genotípicas o virológicas. Se relaciona con la infeccion con Virus de Epstein Barr (VEB): Linfoma de Burkitt africano (endemico). En todos infeccion con latente con VEB linfoma de Burkitt esporádico (no endemico). Una minoría infeccion con VEB Grupo de linfomas agresivos propios de pacientes infectados con VIH. EL 25% infeccion por VEB
- 139. INMUNOFENOTIPO Tumores de células B relativamente maduras que expresan en su superficie IgM, cadenas ligeras monotipicas κ y λ. CD19,20,10 + GENETICA Todos se caracterizan por traslocaciones del gen c-myc situado en el cromosoma 8. Su pareja suele ser el locus de las IgH [t(8;14)] pero también puede ser el locus de las cadeas ligeras κ [t(2;8)] y λ [t(8;22)].
- 140. MANIFESTACIONES CLINICAS. El Linfoma de Burkittafricano la mandibula y preferencia por vísceras abdominales (riñones, ovarios, suprerrenales), Linfoma de Burkitt esporádico, en la región ileocecal y en el peritoneo
- 143. PRONOSTICO. Es un Linfoma agresivo, con respuesta aceptable a ciclos cortos de quimioterapia en dosis altas , con la que muchos pacientes llegan aparentemente a curarse.
- 144. NEOPLASIAS DE CELULAS PLASMATICAS, MIELOMA MULTIPLE Y ENTIDADES AFINES Neoplasias linfoides de las células B totalmente diferenciadas, en células plasmáticas. Causa el 15% de las muertes causadas procesos malignos de los leucocitos Histologia. Secretan Ig monoclonal (componente M) intravascular, y cadenas libres pesadas y libres (proteínas de Bence-Jones), depuradas en orina.
- 145. CLASIFICACION Mieloma multiple (mieloma de células plasmaticas) El mieloma solitario o plasmocitoma LINFOMA LINFOPLASMOCITARIO (Macroglobulinemiade Waldestrom) Amiloidosis primaria asociada a inmunocitos Gammapatiamoonoclonal de significación desconocida (GMSD
- 146. MIELOMA MULTIPLE Neoplasia de células plasmáticas con afectación politópica del esqueleto, y ganglionar y de la piel. Causa el 1% de las muertes en los países occidentales Se observanlesiones osteoliticasmultiples diseminadas por todo el esqueleto a cualquier hueso: columna vertebral 66%, costillas 44%, cráneo 31%, pelvis 28%, femur 24%, clavicula 10%, escapula 10%..Los defectos oseos suelen estar repletos de una masa tumoral roja, blanda, gelatinosa.
- 147. HISTOLOGIA. células plasmáticas normales , o plasmoblastos (cromatina menos densa, un solo nucléolo definido), células multinucleares abigarradas, células en llama (citoplasma rojo vivo), células de Mott (gotas citoplasmáticas de color azul, parecidas a uvas) células con inclusiones (fibrillas, bastocillos, cristalinos, cuerpos de Russell y equivalentes intranucleares, cuerpos de Dutcher)
- 149. GENETICA. Frecuentes deleciones de 13q y reordenamientos de 14q; 30% casosla t(4;14) (p16,3, q32) que afecta la proliferación celular ASPECTOS DIAGNOSTICOS Proteinas de Bence Jones en orina (electroforesis o calenteamiento) la proteína M (inmunoelectroresis o inmunofijacion) Rx.aspecto de un defecto bien delimitado, en sacabocado, que produce en la placa una imagen redonda “en pompa de jabon”
- 150. MANIFESTACIONES CLINICAS Dolor de huesos y fractura patológica Hipercalcemia, secundaria a la resorción del hueso confusionmental, debilidad, aletergamiento, estreñimiento y poliuria Proteinuria de Bence Jones (cadenas ligeras toxicas para los tubulos renales) que favorece las lesiones renales. AmiloidosisAL (producción excesiva de cadenas ligeras de las Ig); infecciones por S. pneumoniae, S. aureus y E. coli; insuficiencia renal. Anemia normocitica y normocromica acompañada (raro) de leucopenia y trombocitopenia moderadas
- 151. LINFOMA LINFOPLASMOCITARIO (Macroglobulinemia de Waldestrom), Neoplasia de células B de los adultos de edad avanzada 60-80 años. Similar a LLC y LLP, pero con diferenciación parcial celular a células plasmáticas. Las células plasmáticas casi siempre secretan IgM monoclonal, que producen el síndrome de hiperviscosidad, Macroglobulinemia de Waldestrom Enfermedad una progresiva incurable. Rara evolución a linfoma de células grandes. Con una mediana de supervivencia de 4 años.
- 152. LINFOMA DE LA ZONA MARGINAL (MALToma) Linfomas de células B en GL, bazo y tejidos extraganglionares, con características morfológicas e inmunofenotipicassimilares Mediana edad Predominancia de las células B parecidas a las células normales de la zona marginal Originados en tejidos afectados por procesos crónicos autoinmunitarios (p.e. Glandulas salivales en el S. de Sjogren y tiroides en la Tiroiditis de Hashimoto) o infecciosos (p.e. estomago con gastritis por Helicobacter) reactivo, inmunitario, policlonal, con evolucion a neoplasia de células B monoclonales
- 153. Figura 4: Endoscopía, estómago – la mucosa presenta difusamente enantema de grado moderado, con discreta nudosidad en la región prepilórica Figura 5: células linfoides infiltrando difusamente la mucosa gástrica con inmunopositividad para el anticuerpo anti-CD20. IHQ, 4X.
- 154. LINFOMAS DE CELULAS DEL MANTO Constituyen el 3%, en EEUU, y entre el 7-9%, en Europa, de los LNH. Es de predominio en hombre, y la edad de 40-60 años. Patrón de la zona de manto, y difuso.
- 157. INMUNOFENOTIPO. CD19, CD20, CD5 + , y CD23 -; niveles moderamente altos de cadenas pesadas Ig de superficie (IgM e IgD). Lo distingue de LLC y LLP GENETICA. Traslocacion (11;14) (70% casos), afecta al locus IgH del cromosoma 14 y el locus BCL1 (PRAD1) del cromosoma 11.
- 158. MANIFESTACIONES CLINICAS. Adenopatias generalizadas y afectación de la MO y el hígado, y esplenomegalia (50% casos). Una minoría presenta síntomas B (fiebre y perdida de peso). 20-40% linfocitosis (<200 000/mm3) Afectación de MO (conglomerados linfoides paratrabecularesno trabeculares), bazo, hígado, ID y colon (poliposislinfomatoide, diseminación multifocal, característica que destaca entre los LNH)
- 159. NEOPLASIAS DE CELULAS PERIFERICAS Y DE CELULAS CITOLITICAS NATURALES (NK) Neoplasias que presentan un fenotipo similar a la de linfocitos T y células NK maduros. Los tumores de células T maduras comprenden alrededor de 15% de los LNH en EEUU y Europa. Los tumores de células NK son comunes en el Extremo Oriente, pero raros en el Hemisferio occidental. Las mas importantes son
- 160. LEUCEMIA/LINFOMA DE CELULAS T DEL ADULTO Neoplasia de las células T CD4+, en pacientes infectados por HTLV-1. Japon. células floridas o en hojas de treboly células multinucleadas parecidas a la células RS Proceso rápidamente progresivo, mortal entre 6 meses y 1 año,
- 161. MICOSIS FUNGOIDE Y SINDROME DE SÉZARY Entidad neoplasica que afecta insidiosamente a las células CD4+ en la piel una mediana de superviviencia de 8-9 años micosis fungoides. células T con un nucleocerebriforme, y pliegues acusados a la membrana nuclear. síndrome de Sézary. eritrodermiaexfoliativa generalizada
- 162. Diagnostico
- 163. Hepatoesplenomegalia Adenopatías múltiples
- 164. Índice internacional de factores pronostico para linfomas no Hodgkin
- 166. Leucemia linfocítica aguda Es un cáncer de crecimiento rápido en el cual el cuerpo produce una gran cantidad de glóbulos blancos inmaduros (linfocitos). Estas células se encuentran en la sangre, la médula ósea, los ganglios linfáticos, el bazo y otros órganos.
- 167. Fisiopatología: La (LLA) conforma el 80% de las leucemias agudas de la niñez y la mayoría de los casos ocurre en niños entre los 3 y los 7 años de edad. Esta enfermedad también se puede presentar en adultos. En la leucemia aguda, las células cancerosas se multiplican rápidamente y reemplazan las células normales. Estas células cancerosas toman el control de partes normales de la médula ósea, causando su insuficiencia. Una persona con leucemia linfocítica aguda tiene mayor probabilidad de sangrar y tener infecciones dado que hay menos células sanguíneas normales.
- 168. La mayoría de los casos de leucemia linfocítica aguda no tiene una causa obvia; sin embargo, lo siguiente puede jugar un papel en el desarrollo de la leucemia: Problemas cromosómicos Radiación Algunos fármacos quimioterapéuticos Toxinas como el benceno
- 169. Morfología El aspecto histológico es caracterizado por arquitectura tisular normal, que ha sido sustituida completamente por linfoblastos de escaso citoplasma y núcleos algo mayores que los de los linfocitos pequeños. En ocasiones la membrana nuclear muestra pliegues profundos que comunican al núcleo, poseen un aspecto retorcido(lobulado).
- 171. Al igual que en otros tumores de elevado índice mitótico como los linfomas de Bukitt, puede verse una imagen en cielo estrellado, debido a los macrófagos benignos que están intercalados.
- 172. Manifestaciones Clínicas Sangrado profuso o prolongado Sangrado nasal Inflamación y sangrado de encías. Dolor o sensibilidad en los huesos Tendencia a la formación de hematomas Fatiga, fiebre, palidez y pérdida involuntaria de peso Dolor articular Infección
- 173. Irregularidades menstruales Petequias Dificultad para respirar (agravada por el ejercicio) Linfoadenopatía Debido al fracaso de la hematopoyesis se presentan cuadros de anemia, neutropenia y trombocitopenia.
- 174. Pronóstico Con quimioterapia enérgica, más del 90% de los niños con LLA consiguen una remisión completa, y dos tercios como mínimo puede considerarse que están curados. Los niños de 2 a 10 años de edad con fenotipo pre-B e hiperploidia, así como los que tienen una translocacion t (12; 21), tienen un menor pronóstico.
- 175. Leucemia linfocítica crónica (LLC) La leucemia linfocítica crónica (LLC) causa un incremento lento en el número de Linfocitos B en la médula ósea. Las células cancerosas se diseminan desde la médula ósea hasta la sangre y también pueden afectar los ganglios linfáticos y otros órganos. Este tipo de leucemia finalmente provoca insuficiencia de la médula ósea y debilita el sistema inmunitario.
- 176. Fisiopatología Se desconoce la razón de este incremento en las células B y no existe un vínculo con la radiación, químicos cancerígenos o virus. Afecta principalmente a los adultos
- 177. Morfología La arquitectura de los ganglios linfáticos esta sustituida por una población celular en la que predominan los linfocitos pequeños de 6-12micras que tienen núcleos redondeados o algo irregulares con cromatina densa y citoplasma escaso. Estos elementos están mezclados con un número variable de otras células mas grandes llamadas prolinfocitos.
- 179. En la LLC, la sangre periférica contiene abundantes linfocitos pequeños, redondos, y de escaso citoplasma. Se observa la afectación de la medula ósea en todos los casos de LLC, adoptando la forma de infiltrados intersticiales o de conglomerados no paratrabeculares de linfocitos pequeños. La pulpa blanca y la pulpa roja del bazo, así como los espacios porta del hígado, suelen estar infiltrados por celular tumorales.
- 180. Manifestaciones Clínicas La mayoría de los pacientes tiene más de 50 años, se ha observado que predomina en el hombre. Generalmente, los síntomas se desarrollan en forma gradual. Muchos casos de LLC se detectan por medio de exámenes sanguíneos de rutina en personas asintomáticas.
- 181. Los síntomas que pueden presentarse abarcan: Hematomas anormales (ocurre en las últimas etapas de la enfermedad) Inflamación de los ganglios linfáticos, el hígado o el bazo (50 a 60%) Sudoración excesiva, sudores fríos Fatiga y fiebre Infecciones recurrentes Pérdida de peso involuntaria Trombocitopenia autoinmunitaria
- 182. Pronóstico La evolución y el pronóstico de la LLC es susceptiblemente variable, y depende principalmente del estadio clínico. En general, la supervivencia media es de 4 a 6 años, pero los pacientes que tienen una masa tumoral mínima pueden vivir 10 años o más.
- 183. Otro factor determinante para la supervivencia del paciente es la tendencia de esta patología a transformarse en neoplasias linfoides más agresivas. Se da en un 15 a 30% de los casos, puede ser de tipo prolinfocitico o de células grandes, suelen tener un mal pronóstico, ya que las personas mueren en menos de un año, después de ser diagnosticado.
- 184. Neoplasias mieloides
- 185. La característica común que engloba a este grupo heterogéneo de neoplasias es su procedencia de un progenitor que normalmente genera células totalmente diferenciadas de la serie mieloide (hematíes, granulocitos, monocitos y plaquetas). Afectan en primer lugar a la medula ósea y, en menor grado, a los órganos hematopoyéticos secundarios (bazo, hígado y ganglios linfáticos).
- 187. Leucemia Mieloide Aguda La LMA afecta principalmente a los adultos y su incidencia es mayor entre los 15 y 39 años de edad, pero se observa de igual manera en niños y adultos mayores. Constituye solo el 20% de las leucemias infantiles. La LMA es bastante heterogénea.
- 188. Fisiopatología Al igual que otras formas de neoplasia, se asocia a alteraciones genéticas adquiridas, o de manera menos común, heredadas, que producen la sustitución de los elementos medulares normales por blastos bastante diferenciados que muestran uno o más rasgos de diferenciación mieloide precoz
- 189. Los precursores mieloides neoplasicos se acumulan en la medula ósea e inhiben al resto de los progenitores medulares normales, reemplazándolos de forma puramente física. El fallo de la hematopoyesis produce anemia, neutropenia y trombocitopenia, son la base de la mayoría de complicaciones observadas en leucemias mieloides agudas.
- 190. Morfología El diagnostico de LMA se basa en el hallazgo de mas de un 30% de blastos mieloides en la médula ósea. Con la tinción habitual de Wright-Giemsa, se puede distinguir los mieloblastos tienen una cromatina nuclear fina, dos a cuatro nucléolos, y un citoplasma más abundante que los linfoblastos. En su citoplasma se encuentran frecuentemente finas granulaciones azurofilas, Peroxidasa-positivas.
- 191. En muchos casos se encuentran también estructuras bacilares teñidas en rojo, llamadas bastoncillos de Auer
- 192. Implicaciones genéticas Al utilizar técnicas especiales de alta resolución para revelar las bandas cromosómicas, se han encontrado alteraciones cromosómicas en el 90% aproximadamente de todos los pacientes con LMA. La LMA que surge de novo en los pacientes sin ningún factor de riesgo suele cursar con translocaciones cromosómicas equilibradas.
- 193. Las LMA que aparecen después de un síndrome mielodisplasico o de contacto con agentes lesivos para el ADN (quimioterapia o radioterapia) suele asociarse a deleciones o monosomias de los cromosomas 5 y 7, y generalmente no tienen translocaciones cromosómicas.
- 194. Manifestaciones clínicas Parecidas a los de leucemias linfoides agudas. La mayoría de los pacientes consultan semanas o pocos meses después de comenzar los síntomas, los cuales están relacionados con la anemia, neutropenia y trombocitopenia, destacando principalmente el cansancio, fiebre y hemorragias cutaneomucosas espontaneas.
- 195. En muchos casos, la diátesis hemorrágica secundaria a la trombocitopenia es el hallazgo clínico y anatómico mas llamativo de la patología. De igual manera se puede observar petequias y equimosis en la piel, así como hemorragias en la superficie serosa de las cavidades corporales y en los revestimientos serosos de las vísceras, especialmente del corazón y los pulmones. Son muy frecuentes las hemorragias en encías y vías urinarias.
- 196. Pronóstico Un 60% de los pacientes aproximadamente consigue una remisión completa con quimioterapia, pero solo un 15 a 30% de ellos sigue sin tener signos o síntomas de la enfermedad 5 años después. Las LMA asociadas a translocaciones de los cromosomas, poseen un mejor pronostico que las asociadas a deleciones o monosomias cromosómicas
- 197. Síndromes mielodisplasicos En los pacientes con SMD, la medula ósea ha sido sustituida parcial o totalmente por la descendencia clonal de una célula madre pluripotencial mutante, que conserva su capacidad para diferenciarse y formar hematíes, granulocitos y plaquetas, pero de una manera que es ineficaz y desordenada.
- 198. Existen dos variedades distintas de SMD: SMD idiopático o primario: Aparece principalmente en pacientes mayores de 50 años y que suele evolucionar insidiosamente. SMD secundario al tratamiento (SMD-t): que es una complicación de un tratamiento anterior con fármacos mielosupresores o con radioterapia, y que suele aparecer 2 a 8 años después de esa exposición.
- 199. Todas las formas de SMD pueden convertirse en una LMA, pero esta transformación se produce con mayor rapidez y frecuencia en los pacientes con SMD-t. Las alteraciones morfológicas características se descubren habitualmente en la médula ósea y en la sangre periférica
- 200. Patogenia La patogenia de los SMD se desconoce, pero se plantea la posibilidad, que los SMD pudieran deberse a una lesión subyacente de las células madre. La forma primaria de los SMD como los SMD-t se acompañan de alteraciones cromosómicas clonales que son similares, entre las que se encuentran la monosomia 5 y la monosomia 7, las deleciones de 5q y 7q, la trisomia 8 y las deleciones de 20q.
- 201. Morfología El hallazgo más característico es la diferenciación desordenada (displasica) de las tres líneas celulares: eritroide, mieloide y megacariocitica. Se manifiesta por tres elementos: Sideroblastos en anillo Maduración megaloblastoide Fenómenos de “gemación” nuclear
- 202. Sideroblastos en anillo, unos eritroblastos cuyas mitocondrias cargadas de hierro aparecen formando granulaciones perinucleares en las muestras de biopsia o en los frotis por aspiración de la medula ósea, teñidos con azul de Prusia.
- 203. Maduración megaloblastoide, semejante a la observada en el déficit de vitamina B12 y de folato.
- 204. Fenómenos de “gemación” nuclear, que ofrecen el aspecto de núcleos deformados y, a menudo, de contornos polipoides. Los neutrófilos tienen a veces pocas granulaciones secundarias, o bien muestran granulaciones tóxicas y cuerpos de Dohle.
- 205. Con frecuencia se observan células pseudo-Pelger-Huet(neutrófilos cuyos núcleos tienen solo dos lóbulos). Son muy típicos también los megacariocitos con un solo lóbulo nuclear o con muchos núcleos separados (megacariocitos con “cabeza” de peón).
- 206. Manifestación clínica El SMD primario afecta principalmente a personas mayores de 60 años. Igual que en la leucemia aguda, los pacientes con este proceso consultan por debilidad, infecciones y hemorragias, manifestaciones todas debidas a la pancitopenia. La mitad aproximadamente de los pacientes no tiene síntomas y la enfermedad se descubre casualmente al hacer un análisis de sangre.
- 207. Pronóstico La mediana de la supervivencia del SMD primario varía entre 9 y 29 meses, pero algunos pacientes pertenecientes a los grupos de buen pronóstico pueden vivir 5 años o más. En general, la evolución hacia una LMA se produce en un 10 a 40% de los casos y suele acompañarse de la aparición de nuevos cambios citogenéticos clonales.
- 208. El pronóstico es mucho más sombrío en los pacientes con SMD-t, cuya mediana de supervivencia es solo de 4 a 8 meses. En ellos, las citopenias suelen ser mucho más profundas que en el SMD primario, y muchos casos evolucionan rápidamente hacia LMA. La opciones terapéuticas del SMD son escasas, en pacientes jóvenes, el trasplantes alogénico de medula ósea ofrece ciertas posibilidades de lograr la recuperación de la hematopoyesis normal y de prolongar la supervivencia.
- 209. Procesos Mieloproliferativos Crónicos (PMC) Leucemia Mieloide crónica Policitemia Vera Trombocitosis esencial Mielofibrosis con metaplasia mieloide
- 210. El elemento que sufre la transformación neoplásica es una célula progenitora pluripotencial capaz de formar hematíes, plaquetas, granulocitos y monocitos maduros. La única excepción es la leucemia mieloide crónica (LMC), en lo que parece estar afectada la célula madre pluripotencial capaz de producir células linfoides y mieloides.
- 211. Al igual que la LMA, las células neoplásicas y su descendencia inundan la medula ósea y suprimen las células progenitoras normales residuales; sin embargo, los PMC se distinguen en que, al principio, no está afectada la diferenciación final. Hipercelularidad medular y aumento de la hematopoyesis, acompañada con frecuencia de valores elevados de los recuentos en sangre periférica.
- 212. Las células madre neoplásicas son capaces de circular y alojarse en los órganos hematopoyéticos secundarios, especialmente en el bazo, donde producen hematopoyesis extramedular.
- 213. El resultado es que todos los PMP crónicos producen esplenomegalia de intensidad variable. Además, suelen terminar en una “fase de agotamiento” caracterizada por fibrosis medular y citopenias hemoperifericas.
- 214. 1. Leucemia mieloide crónica. Esta enfermedad se da principalmente en los adultos con edades comprendidas entre los 25 y 60 años, siendo máxima su incidencia en los decenios cuarto y quinto de la vida. Herencia: La LMC se distingue de otros PMP crónicos por una alteración molecular característica: una traslocaciónque afecta al gen BCR del cromosoma 9 y al gen BCL del cromosoma 22.
- 215. Morfología A diferencia de la medula ósea normal (50% células/50% grasa), el 100% de la medula en LMC suele estar formada por células, la mayor parte de las cuales son precursores granulociticos en fase de maduración. Con frecuencia se observa aumento del número de megacariocitos, que muchas veces son formas pequeñas displasicas.
- 217. Como hallazgo característico se observan, de forma desperdigada, histiocitos de depósito con citoplasma azul marino (histiocitos azul marino), que también se encuentran en otros procesos con aumento del recambio celular de la medula ósea.
- 218. Entre los elementos circulantes predominan los neutrófilos, metamielocitos y mielocitos, y menos de un 10% de mieloblastos. También son frecuentes la eosinofilia y basofilia hemoperifericas, y hasta un 50% de los pacientes tiene Trombocitosis en las primeras fases de la enfermedad.
- 219. Manifestaciones Clínicas Tiene un comienzo insidioso y los primeros síntomas son bastante inespecíficos como cansancio, debilidad, pérdida de peso y anorexia. En ocasiones, el primer síntoma es una sensación de pesadez en el abdomen, causada por esplenomegalia.
- 220. Un dato de laboratorio de bastante utilidad, propio de LMC es la ausencia casi absoluta de fosfatasa alcalina leucocitaria. El único trastorno hematológico que se acompaña de fosfatasa alcalina leucocitaria baja es la hemoglobinuria paroxistica nocturna, que no tiene ninguna semejanza con la LMC en otros aspectos y que, por tanto, se distingue fácilmente.
- 221. Pronostico La LMC evoluciona de forma lentamente progresiva, e incluso sin tratamiento, puede esperarse una mediana de supervivencia de 3 años. Tras un periodo variable, de 3 años en promedio, el 50% aproximadamente de los pacientes entra en una fase acelerada, caracterizada por la falta gradual de respuesta al tratamiento junto con anemia y trombocitopenia crecientes, y a veces con intensa basofilia en sangre periférica.
- 222. En la fase estable, se puede controlar con agentes quimioterapéuticos en dosis bajas, aunque estos no impiden la evolución progresiva hacia una crisis blastica. Alrededor del 75% es curado cuando se recibe el donante adecuado. También se ha utilizado métodos de inhibición a las células progenitoras de LMC como el interferon–alfa, lo que permite que la estirpe de células madre normales repueblen la medula ósea.
- 223. Policitemia VERA (PVC) La proliferación medular excesiva se refleja en la sangre periférica por eritrocitosis(Policitemia), granulocitosis y trombocitosis, pero la alteración responsable de casi todos los síntomas es el aumento absoluto de la masa eritrocitaria.
- 224. Morfología Hay hipercelularidad medular, aunque es frecuente observar algo de grasa residual. El aumento de los progenitores eritroides puede ser bastante sutil, y suele acompañarse de mayor número de megacariocitos y de los precursores granulociticos en maduración.
- 225. En las primeras fases evolutivas de la PCV, cuando la hematopoyesis extramedular es mínima, suele haber ligeras organomegalias debidas en gran parte a congestión. Usualmente en frotis de sangre periférica se encuentra un aumento de basofilos y plaquetas grandes.
- 226. En la fase tardía de la enfermedad, la medula ósea puede evolucionar a la fase de “agotamiento”, que se caracteriza por una fibrosis que ocupa los espacios intertrabeculares y que desplaza a las células hematopoyéticas.
- 227. Manifestaciones clínicas Aparece insidiosamente en personas que sobrepasan la media edad (inicio a los 60 años aproximadamente). La mayoría de los síntomas tienen relación con el aumento de la masa eritrocitaria, que se manifiesta con hematocrito elevado, con un aumento del volumen total de sangre
- 228. Por tanto, los pacientes tienen un aspecto algo cianótico, debido a que la sangre desoxigenada se estanca en los vasos periféricos. Cerca del 70% de los pacientes son hipertensos y, a menudo, se quejan de cefalea, mareos y síntomas gastrointestinales. Prurito intenso y abundantes ulceras pépticas, ambas manifestaciones podrían deberse al aumento en la liberación de histamina por los basofilos.
- 229. Pronóstico Pacientes no tratados mueren por hemorragias o por trombosis, en el plazo de meses luego del diagnostico de la enfermedad. Sin embargo, si la masa eritrocitaria se mantiene cerca de sus niveles normales por medio de sangrías, se puede conseguir una mediana de supervivencia de 10 años.
- 230. Al prolongarse la vida con el tratamiento, se ha observado que la evolución natural de la PCV implica el paso gradual a una fase de agotamiento, durante la cual aparecen manifestaciones clínicas y anatómicas de Mielofibrosis con metaplasia mieloide (cerca del 15 a 20% de los pacientes sufren esta transformación al plazo de 10 años).
- 231. Trombocitosis esencial. Es la forma menos frecuente de PMC, se produce una excesiva proliferación y producción celular de elementos megacariocíticos, y los pacientes presentan cifras de plaquetas superiores a 600,000/mm3
- 232. Las principales manifestaciones clínicas son las trombosis y las hemorragias, que dependen de las alteraciones cualitativas y cuantitativas de las plaquetas. Proceso insidioso con largos periodos asintomáticos acompañados de vez en cuando por crisis tromboticaso hemorrágicas. La mediana de supervivencia es de 12 a 15 años.
- 233. Mielofibrosis con metaplasia mieloide La característica principal que diferencia a esta patología de otros PMP crónicos, es la precocidad con que evoluciona hacia fibrosis medular (Mielofibrosis), que es idéntica histológicamente a la fase de agotamiento de las PCV y otros PMC.
- 234. Fisiopatología: Son el resultado del extenso deposito de colágeno elaborado por los fibroblastos no neoplasicos de la medula ósea. Esta fibrosis desplaza a los elementos hematopoyéticos, incluidos las células madres, y parece deberse a la liberación inadecuada de factores fibrógenos por los megacariocitos neoplásicos.
- 235. Se supone que intervienen dos factores: el factor de crecimiento derivado de las plaquetas (PDGF) y el TGF-B(factor de crecimiento transfomante); ambos son sintetizados por megacariocitos, y liberados a tejidos circundantes. Estos factores son mitogenos de los fibroblastos, además el TGF-B es precursor de fibrosis y angiogenesis(procesos característicos en Mielofibrosis).
- 236. Morfología: Hipercelularidad medular con aumento de todas las líneas celulares que están en fase de maduración. Presencia de megacariocitos grandes, displasicos y forman conglomerados anormales.
- 237. En las últimas etapas evolutivas de la enfermedad, gran parte de la medula fibrosa puede haberse convertido en hueso, un cambio que se denomina osteoesclerosis.
- 238. Debido a la hematopoyesis extramedular hay un agrandamiento de hígado, ganglios linfáticos, y especialmente bazo. En Mielofibrosis bien desarrollada, la sangre periférica muestra una serie de hallazgos característicos: Desestructuración fibrosa, aparición de precursores eritroides y granulociticos en la sangre periférica (leucoeritroblastosis), se observan también hematíes en lagrima (dacriocitos).
- 239. Manifestaciones clínicas Es muy raro en personas menores de 60 años, excepto cuando va precedida de PCV y LMC. Suele manifestarse por anemia progresiva o por aumento notable del tamaño del bazo, los síntomas inespecíficos son cansancio, pérdida de peso y sudores nocturnos.
- 240. Debido al elevado recambio celular, el cuadro puede complicarse con hiperuricemia y gota secundaria. Para el diagnostico es esencial extraer una biopsia de medula ósea donde se pueda detectar el depósito de reticulina, o una fibrosis avanzada.
- 241. Pronóstico La evolución de la enfermedad es difícil de predecir. En distintas series, la mediana de la supervivencia varía entre 1 y 5 años. La vida está amenazada por las infecciones, episodios de trombosis, hemorragias relacionadas con alteraciones de las plaquetas y, en un 5 a 20% de los casos, por la transformación en una LMA.
- 242. HISTIOCITOSIS
- 243. Definición: Grupo de enfermedades que se caracterizan por la proliferación de macrófagos en diferentes órganos y sistemas.
- 244. Hay tres clases importantes de histiocitosis: Histiocitosis de las células de Langerhans, también llamada histiocitosis X Síndrome de histiocitosis maligna (ahora conocido como linfoma de células-T) Histiocitosis de células no-Langerhans (también conocido como síndrome hemofagocítico)
- 245. Histiocitosis de Células de Langerhans Constituye proliferaciones clonales de células dendríticas presentadoras de los antígenos, que se observan fácilmente en la piel en condiciones normales, pero que también pueden encontrarse en otros órganos.
- 246. Antiguamente, estos procesos se llamaban Histiocitosis X y se dividían en tres grupos: la enfermedad de Latterer-Siwe, la enfermedad de Hand-Shuller-Christian y el granuloma eosinófilo. Actualmente se supone que estas tres entidades son formas distintas de manifestación de un mismo proceso básico.
- 247. Causas, incidencia y factores de riesgo se trata realmente de un fenómeno autoinmunitario, en el cual células inmunitarias anómalas atacan al cuerpo, en lugar de combatir las infecciones. Las células inmunitarias extra pueden formar tumores, que pueden afectar diversas partes del cuerpo, incluyendo los huesos, el cráneo y otras áreas. Algunas formas de la enfermedad son genéticas.
- 248. Afecta a aproximadamente 1 de cada 200.000 personas al año Con mayor frecuencia se presenta en niños de 1 a 15 años, con una tasa que alcanza su punto máximo entre los niños de 5 a 10 años
- 249. Histológicamente se observan células tumorales dendríticas presentando antígenos y expresan HLA-DR y CD1a.
- 250. Citoplasma abundante, y a menudo vacuolado, y núcleos ovalados vesiculosos o con muescas. Citoplasma con cuerpos HX (granulaciones de Birbeck) las cuales forman estructuras tubulares parecidas a bastoncillos pentalaminares, con una periodicidad característica y, a veces, un extremo final dilatado (aspecto de raqueta de tenis).
- 251. Las manifestaciones clínicas más frecuentes son: lesiones cutáneas parecidas a una erupción seborreica (afecta a la cara anterior del tronco, espalda y cuero cabelludo) Hepatoesplenomegalia Adenopatías lesiones pulmonares lesiones osteolíticas destructivas
- 253. Histiocitosis de células de Langerhans unifocal y multifocal Acumulaciones expansivas y erosivas de células de Langerhans dentro, generalmente, de la cavidad medular de los huesos. Histológicamente: Histiocitos mezclados en proporciones variables con eosinófilos, linfocitos, células plasmáticas y neutrófilos.
- 254. Lesiones Unifocales: Afectan el esqueleto. Pueden causar dolores espontáneaos o ser asintomáticas y provocados, a veces, por fracturas espontáneas. Puede curar espontáneamente o después de una irradiación o extirpación local. Histiocitosis multifocal de las células de Langerhans: Los infiltrados de células de Langerhans provocan adenopatías ligeras, hepatomegalia y esplenomegalia. El conjunto de osteolisis de la bóveda craneal, diabetes insípida y exoftalmos se conoce como tríada de Hand-Shuller-Christian. En ciertos casos hay regresión espontánea de este proceso; sino, puede tratarse con quimioterapia.
- 255. Complicaciones Las complicaciones pueden abarcar: Fibrosis pulmonar intersticial difusa Neumotórax espontáneo Los niños también pueden presentar: Anemia causada por la diseminación de tumores a la médula ósea Diabetes insípida Problemas del pulmón que llevan a insuficiencia pulmonar Problemas con la hipófisis que conducen a retraso en el crecimiento.
- 256. El Bazo
- 257. El bazo está situado en el hipocondrio izquierdo, inmediatamente debajo del diafragma, encima del riñón izquierdo y del colon descendente y detrás del fondo gástrico Normalmente el bazo del adulto pesa unos 150 g y mide 12 cm de largo por 7 cm de ancho y 3 cm de grueso.
- 259. Histología Es un órgano que está en capsulado, dicha capsula se compone de tejido conectivo denso cubierto de mesotelio escamoso, además de fibras colágenas y elasticas abundantes; incluye celulas del musculo liso ya que el bazo presenta una cierta contractilidad.
- 261. El bazo normal Funciones: Filtrar la sangre circulante de todas sus partículas o cuerpos extraños y participar en la respuesta inmunitaria contra todos los antígenos vehiculados por la sangre.
- 262. Conociendo la función esplénica normal, estas funciones pueden separarse en cuatro grupos: La filtración de los elementos formes de la sangre defectuosos por los fagocitos de los cordones del bazo El bazo es un importante órgano secundario del sistema inmunitario. La malla reticular de las vainas linfáticas atrapa a los antígenos, permitiendo que se pongan en contacto con los linfocitos efectores. El bazo es una fuente de células linforreticulares y a veces de células hematopoyéticas Gracias a su vascularización y a su función fagocitaria el bazo constituye también un órgano de reserva y de depósito de los componentes sanguíneos
- 263. Patología del Bazo El bazo rara vez es el lugar primario de la enfermedad. Cuando está afectado por una enfermedad sistémica aumenta de tamaño, por lo tanto la esplenomegalia es la manifestación más importante de este órgano.
- 264. ESPLENOMEGALIA El aumento del tamaño del bazo es a veces una pista diagnóstica importante que denuncia la existencia de una enfermedad subyacente, pero la propia esplenomegalia puede causar problemas.
- 266. Manifestaciones Clínicas Sensación de pesadez Al comprimir el estómago provoca molestias después de comer Dolor en cuadrante superior izq. Dolor a la palpación Masa abdominal
- 267. Por su función de depósito puede causar: Hiperesplenismo: que se caracteriza por la tríada de: 1) Esplenomegalia 2) Reducción de dos o mas de los elementos sanguíneos 3) Corrección de las citopenias
- 268. Las esplenomegalias más significativas son: Esplenitis Aguda inespecífica Esplenomegalia congestiva Infartos Esplénicos
- 269. Esplenitis Aguda Inespecífica Se produce por un aumento del bazo denominado tumor esplénico, en cualquier infección hematógena. La reacción esplénica inespecífica en estas infecciones puede deberse no solo a los agentes microbianos, sino también a los productos de la enfermedad inflamatoria.
- 270. Morfología MACROSCOPICAMENTE El bazo aumenta de peso de 200 a 400 g. La sustancia esplénica está fluidificada y suele escurrirse por la superficie de corte. MICROSCOPICAMENTE el principal cambio es la congestión aguda de la pulpa roja, que puede invadir y borrar prácticamente a los folículos linfoides.
- 271. Esplenomegalia congestiva La congestión venosa crónica o persistente puede aumentar el tamaño del bazo, produciendo una esplenomegalia congestiva, la causa mas frecuente de esplenomegalia congestiva son las diversas formas de cirrosis hepática, especialmente la alcoholica.
- 273. El órgano es firme y consistente, la superficie de corte es de aspecto carnoso de color gris-rojizo-oscuro.
- 274. La capsula aumenta de grosor y es fibrosa y
- 276. Puede haner focos de hemorragia con depósitos de hemosiderina en los histiocitos.
- 279. Fibromas
- 280. Osteomas
- 281. Condromas
- 282. Linfangiomas
- 285. Enfermedad de Hodgkin: caracterizada por la proliferación neoplasica de una forma atípica de células linfoide denominada RS
- 287. Anomalias congénitas Ausencia de bazo Hipoplasia Bazos accesorios
- 292. Hiperplasia del Timo El término hiperplasia tímica se aplica en realidad al aspecto que ofrecen los folículos linfoides situados dentro del timo y que dan lugar a lo que se llama hiperplasia folicular del timo. Estos folículos linfoides son iguales a los que se encuentran en los ganglios linfáticos, tienen centros germinales y contienen células foliculares dendríticas y linfocitos B, que escasean en el timo normal.
- 293. Timomas Son varias las neoplasias que pueden aparecer en el Timo: Tumores de células Germinales, linfomas, enfermedad de Hodgkin y Carcinoides, entre otros, pero el nombre de timoma debe restringirse a los tumores del timo formados por células epiteliales
- 295. Timoma Benigno o Encapsulado: citológica y biológicamente benigno.
- 296. Timoma Maligno.
- 297. Tipo I, llamado también timoma invasor: Es benigno citológicamente, pero agresivo biológicamente y capaz de producir invasión local y, raras veces, diseminación a distancia.
- 303. Son tumores propios del adulto mayores de 40 años raros en los niños Igual en hombres que en mujeres Crecen en mediastino anterosuperior, a veces lo hacen en el cuello, tiroides, hilio pulmonar o en otros lugares poco frecuentes en el mediastino posterior
- 304. Morfología Macroscópicamente los timomas son masas lobuladas, de consistencia firme, color blanco grisáceo y de 15 a 20 cm de eje mayor. La mayoría parecen estar encapsulados, pero casi el 20 a 25% de ellos ofrecen una penetración evidente de la capsula con infiltración de las estructuras y tejidos que rodean al timo.
- 306. I Encapsulado totalmente, sin objetivarse invasión macro o microscópica de la cápsula.
- 307. II Invasión macro y microscópica de la cápsula tumoral y diseminación a la grasa y/o pleura mediastínica.
- 308. III Invasión macroscópica de otros tejidos adyacentes como el pericardio, grandes vasos o parénquima pulmonar. IV a Metástasis pericárdicas y/o pleura mediastínica.