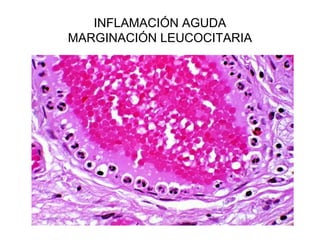
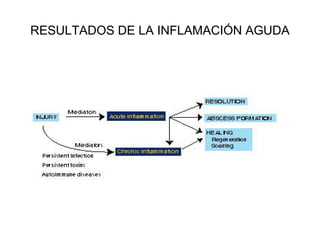
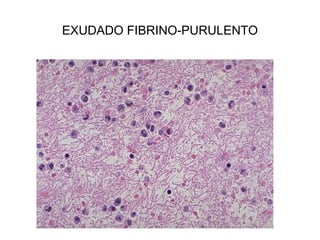
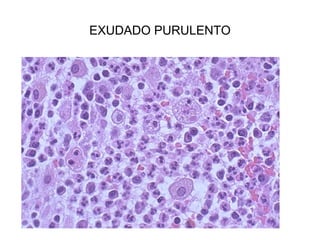
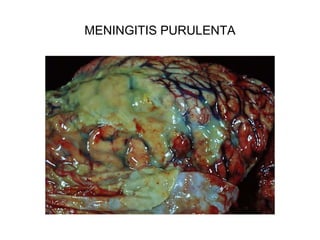
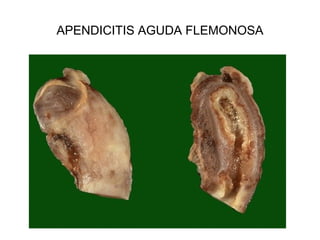
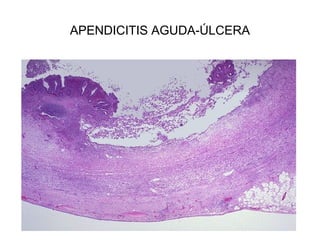
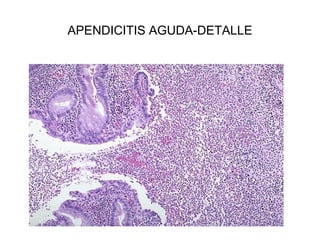
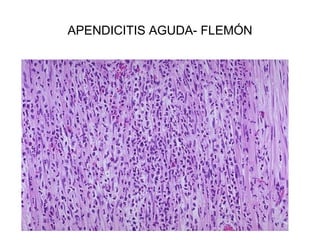
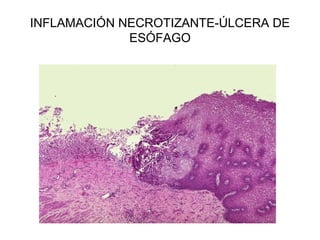
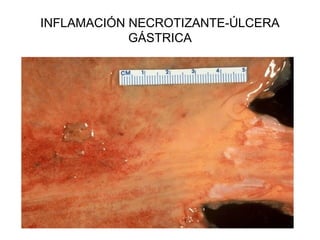
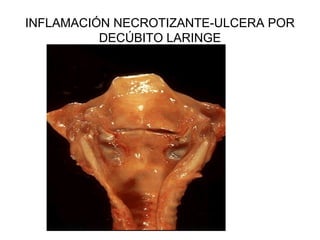
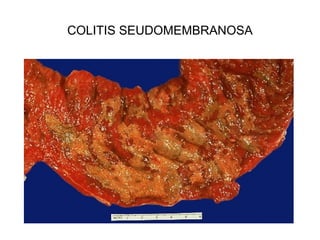
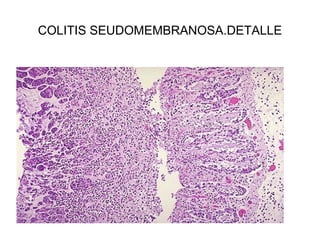
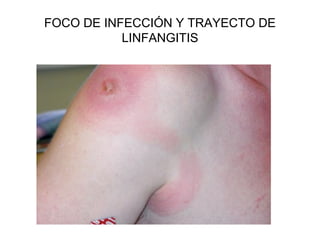
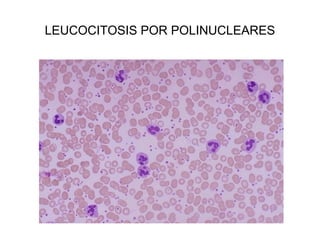
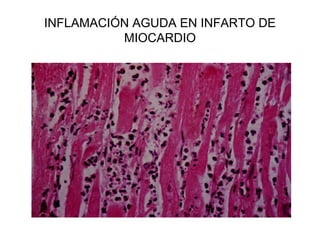
Este documento describe los cambios vasculares, celulares y mediadores químicos involucrados en la inflamación aguda. Explica que la inflamación aguda causa vasodilatación y aumento de la permeabilidad vascular, lo que lleva a la marginación y migración de leucocitos al sitio de inflamación. También involucra la liberación de mediadores como histamina, prostaglandinas y citoquinas que regulan estos procesos inflamatorios.













































![Membranas Celulares 2º Bachillerato [Modo De Compatibilidad]](https://cdn.slidesharecdn.com/ss_thumbnails/membranascelulares2bachilleratomododecompatibilidad-091130170425-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)








































