
Descargar como PDF, PPTX





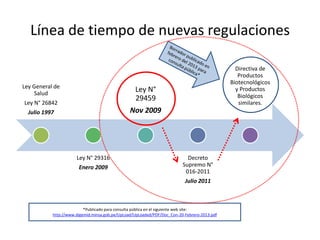

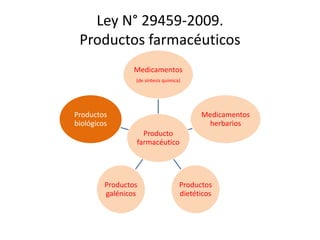
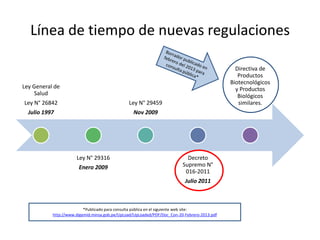


















Este documento presenta un resumen de las nuevas regulaciones de productos biotecnológicos en el Perú. Se describe la línea de tiempo de las leyes y decretos que han establecido nuevos requisitos para la aprobación de estos productos desde 1997 hasta la actualidad. Finalmente, se resume el anteproyecto de Directiva de Productos Biotecnológicos, el cual establecerá los requisitos específicos para la aprobación de estos productos una vez sea publicado.









































































































