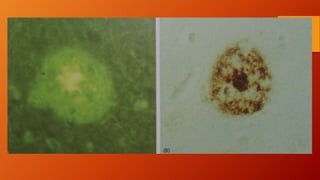
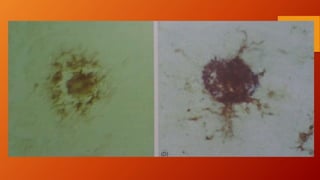
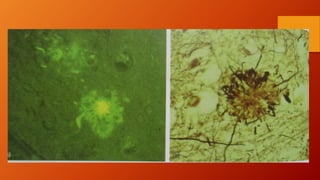
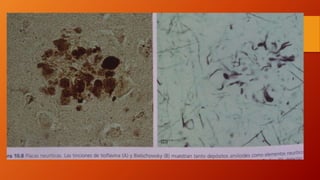
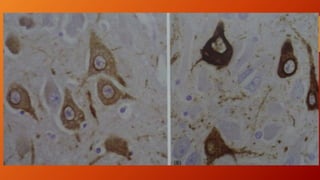
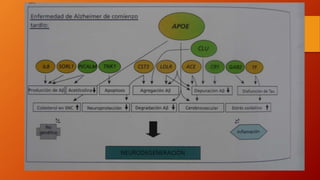
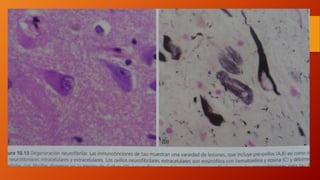
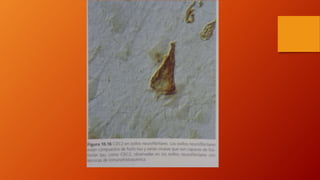
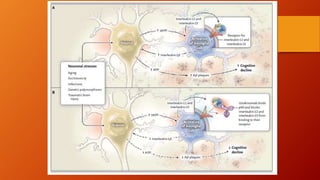
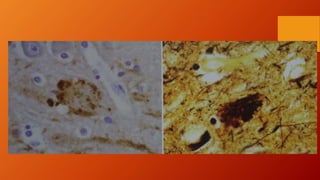
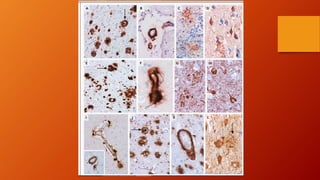
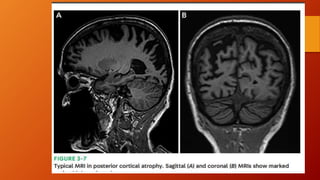
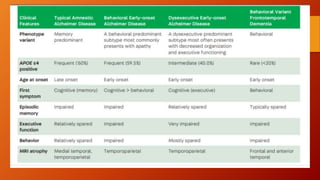
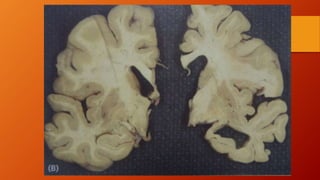
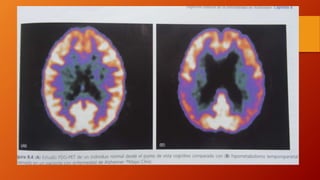
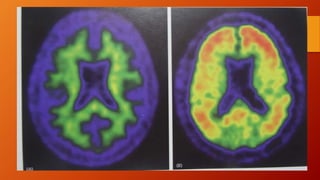
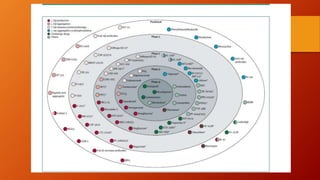
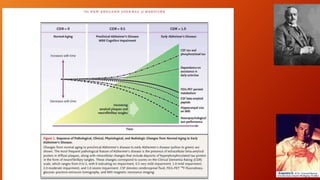
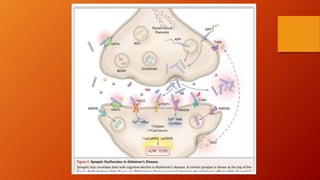
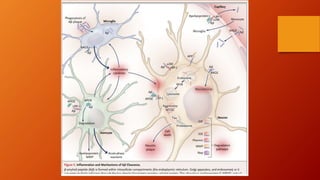
Este documento describe la enfermedad de Alzheimer, incluyendo su fisiopatología, factores de riesgo, manifestaciones clínicas, diagnóstico y tratamiento. Explica que la EA se debe principalmente a la acumulación anormal de las proteínas beta amiloide y tau en el cerebro, lo que causa daño y muerte neuronal. El diagnóstico se basa en criterios clínicos, aunque los biomarcadores como imágenes cerebrales y análisis de liquido cefalorraquídeo pueden ayudar. El