Descargado 219 veces








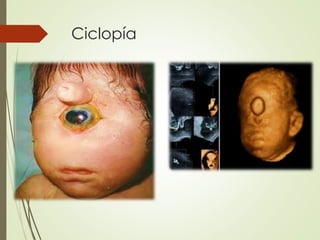




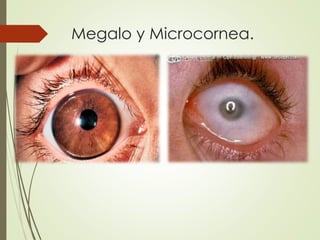









Este documento describe varias anomalías oculares congénitas, incluyendo anoftalmia, microftalmia, nanoftalmia, criptoftalmia, ciclopía, coloboma, megalocornea, microcornea, embriotoxon posterior, anomalía de Axenfeld, anomalía de Rieger, anomalía de Peters y aniridia. Define cada anomalía y explica sus características clínicas.

































![[Embriología] - Formación de Oído y Ojo](https://cdn.slidesharecdn.com/ss_thumbnails/embriologa-seminarioformacindeojoyodo-111016101513-phpapp02-thumbnail.jpg?width=640&height=640&fit=bounds)

































