Descargado 95 veces

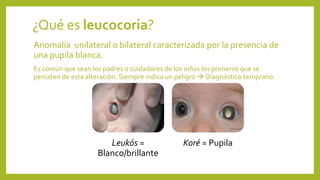
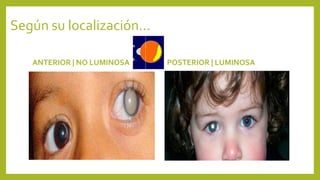


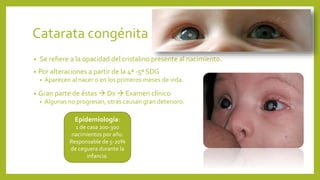


















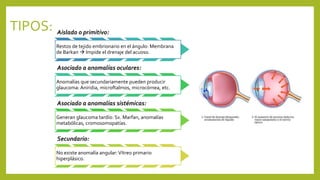





Este documento describe varias anomalías oculares pediátricas, incluyendo: 1) Leucocoria, que es una pupila blanca que puede indicar catarata, retinoblastoma u otras masas oculares; 2) Retinopatía del prematuro, que afecta a bebés prematuros y puede causar ceguera si no se trata; 3) Retinoblastoma, un tumor ocular maligno común en niños menores de 5 años; y 4) Glaucoma congénito, una elevación de la presión ocular que puede caus











































































