Descargado 30 veces

























































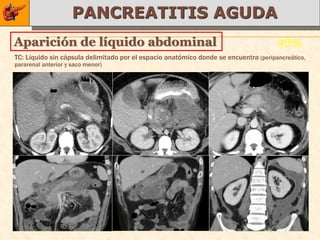








Este documento trata sobre tres temas principales de endocrinología y metabolismo aplicados a la nutrición y dietética: la cirrosis hepática, la esplenomegalia y anemias. La cirrosis hepática es una enfermedad crónica e irreversible que puede deberse a diversas causas como el alcoholismo o hepatitis virales. La esplenomegalia puede ser masiva, moderada o leve y estar asociada a desórdenes mieloproliferativos u otras enfermedades. Finalmente, el documento describe las anemias por defici





























![Cirrosis completo.pp.ppt [reparado]](https://cdn.slidesharecdn.com/ss_thumbnails/cirrosiscompleto-pp-pptreparado-120619132949-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)

























































