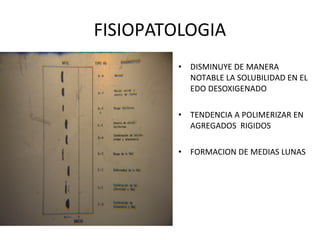
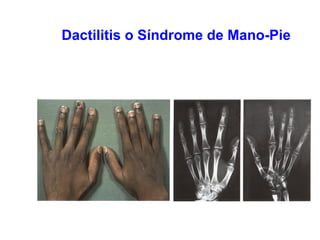
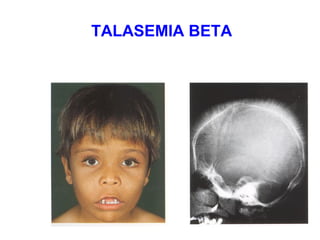
Este documento describe la drepanocitosis y las talasemias. La drepanocitosis es causada por una mutación en la cadena beta de la hemoglobina que hace que se polimerice cuando los niveles de oxígeno son bajos, lo que causa daño a los eritrocitos. Los síntomas incluyen anemia, crisis vaso-oclusivas, infecciones y daño a órganos. El tratamiento incluye medidas preventivas, hidroxiurea y trasplante de médula ósea. Las talasemias son trastornos



































































































![Anemia de células_falciformes[1]](https://cdn.slidesharecdn.com/ss_thumbnails/anemiadeclulasfalciformes1-150521032223-lva1-app6892-thumbnail.jpg?width=640&height=640&fit=bounds)




