Descargar como PDF, PPTX



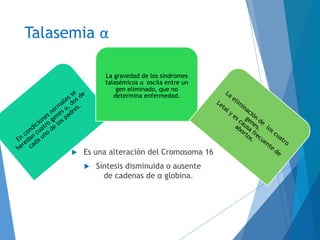





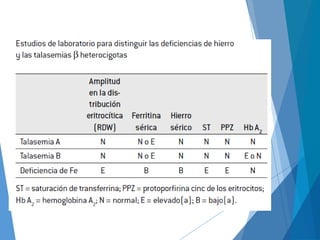
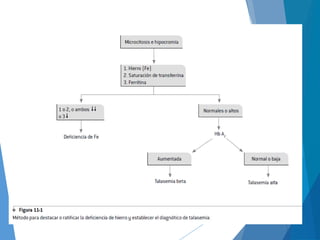


Este documento resume las características principales de las talasemias. Explica que las talasemias son desórdenes congénitos causados por una deficiencia en la síntesis de las cadenas alfa o beta de la hemoglobina. Se clasifican en talasemia alfa y beta. La talasemia alfa implica una síntesis disminuida de cadenas alfa, mientras que la talasemia beta implica una síntesis deficiente de cadenas beta. Las formas pueden ser menores, que son asintomáticas, o mayores, que











































































































