Descargado 44 veces


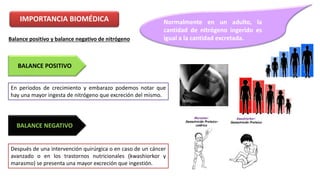
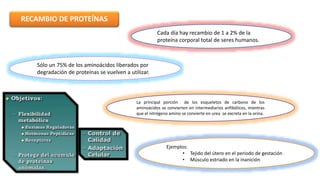
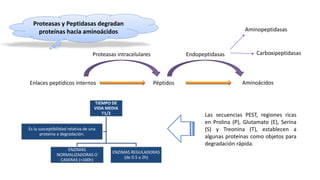
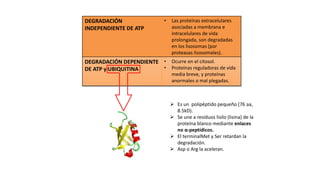
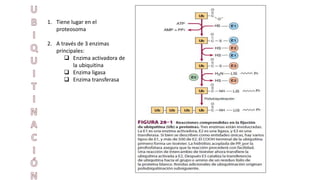

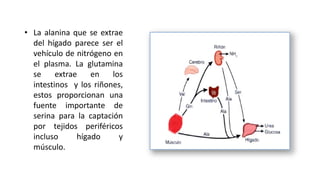
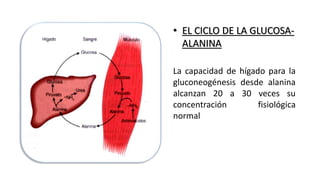

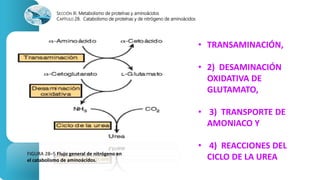
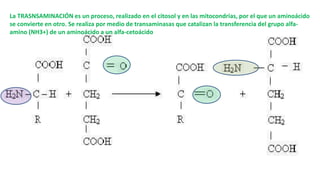
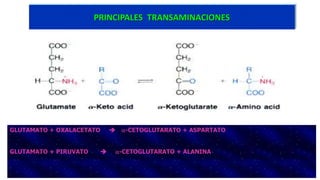
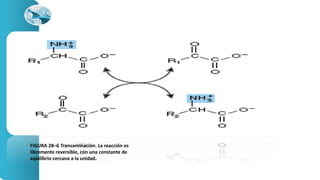
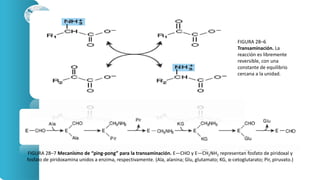
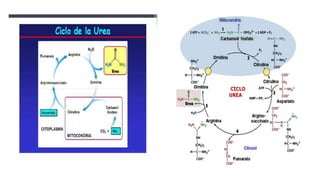
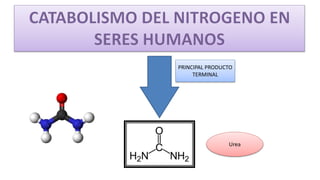
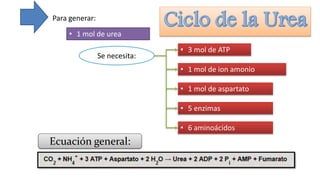
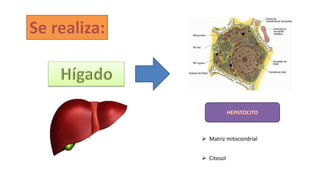
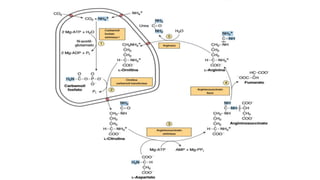
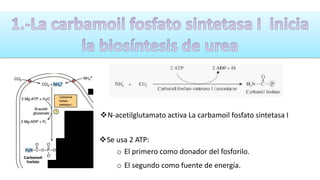
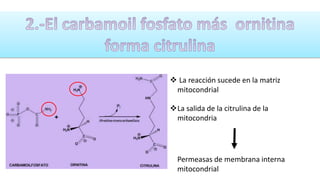
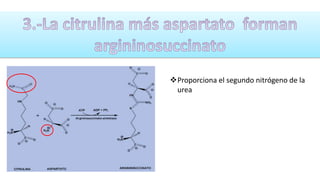
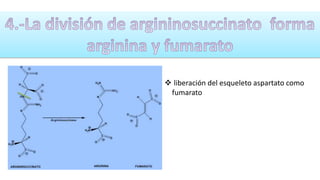
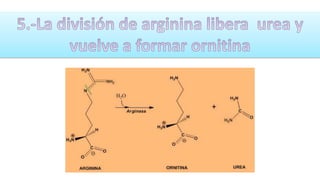
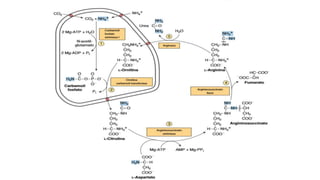
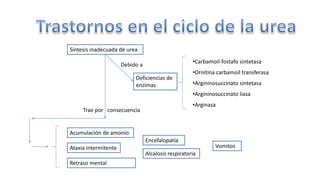
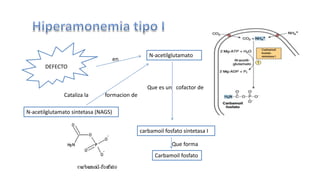
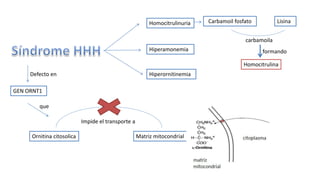
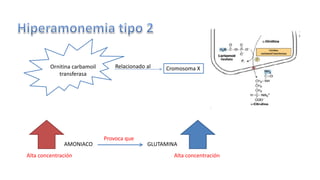
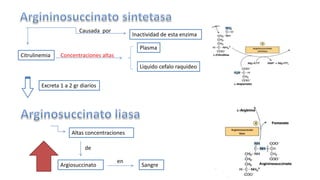
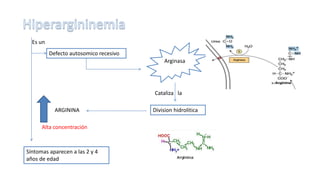


El documento describe los procesos de degradación y recambio de proteínas en el cuerpo humano. Normalmente, la cantidad de nitrógeno ingerido es igual a la excretada, pero en periodos de crecimiento o embarazo hay más ingesta que excreción. Después de cirugías o enfermedades, ocurre lo contrario. Las proteínas se degradan en aminoácidos a través de enzimas proteolíticas y peptidásicas. El principal producto de desecho del nitrógeno es la urea, cuya síntesis requ



































































