Descargado 1207 veces















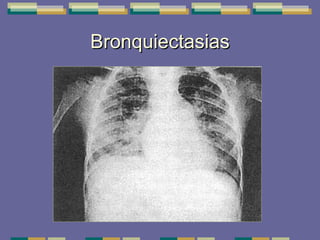
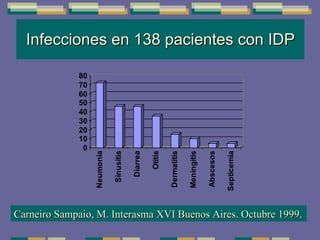
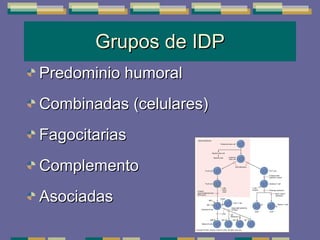




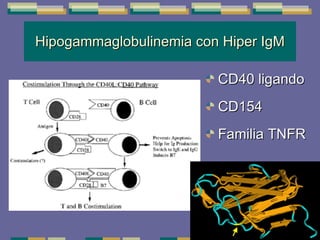
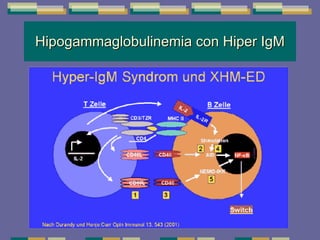







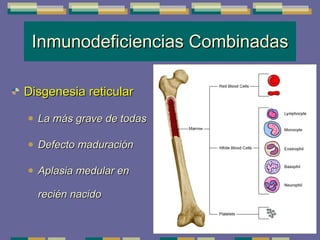

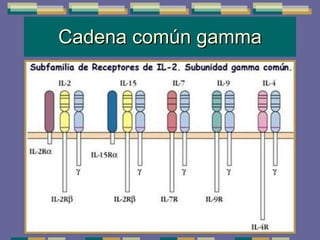
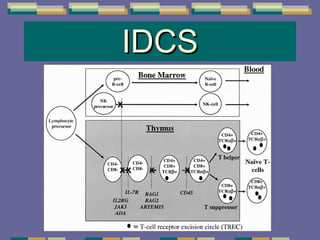
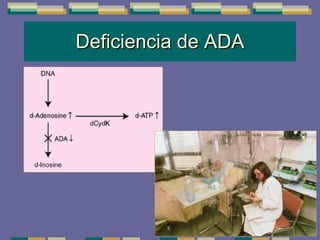




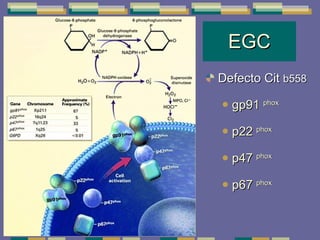
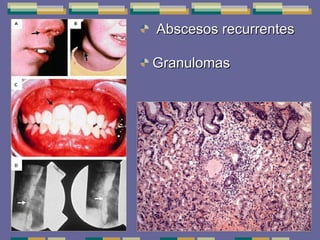
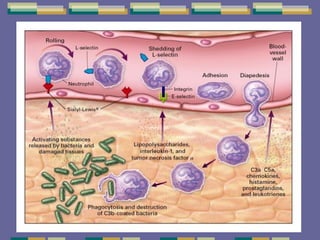

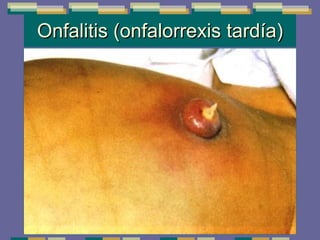
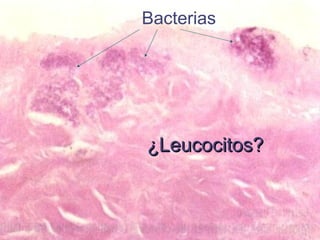


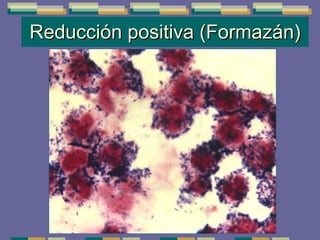




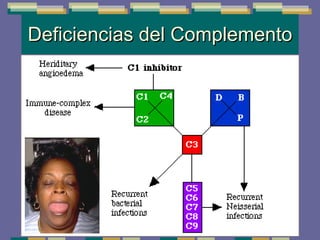
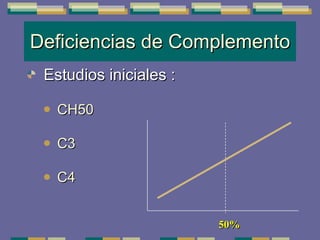
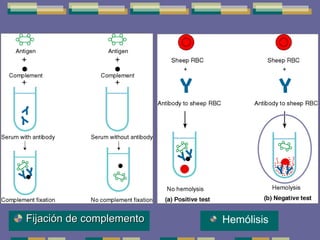



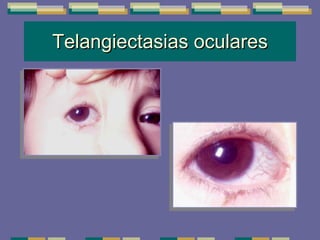
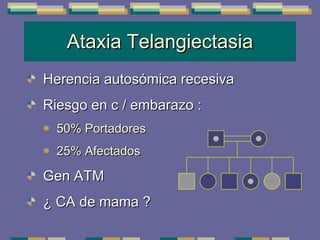



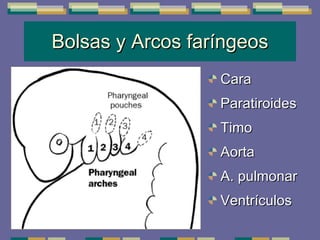

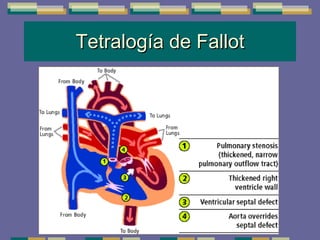







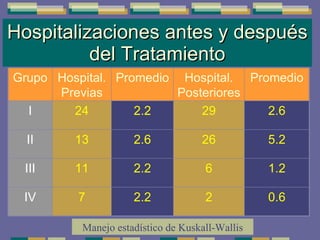

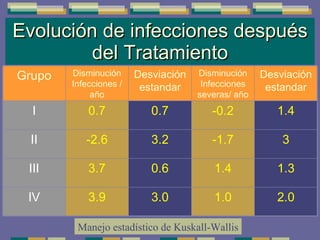

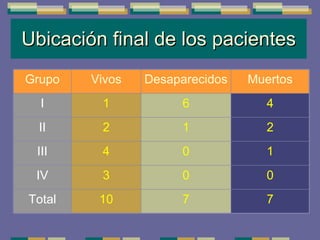


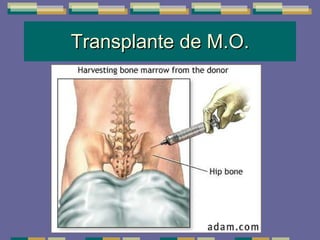
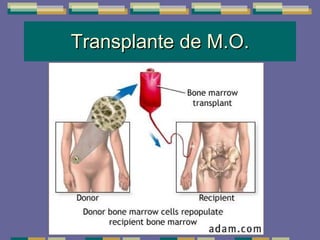
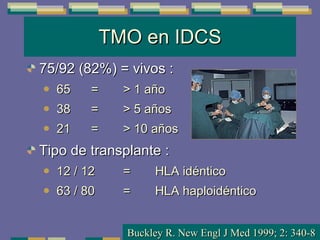

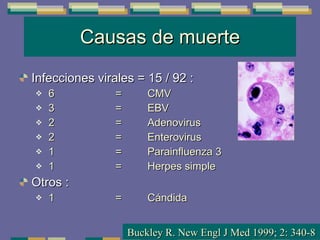
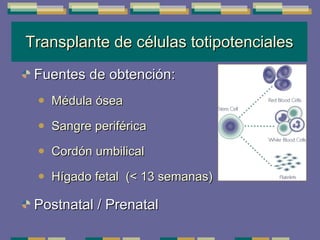



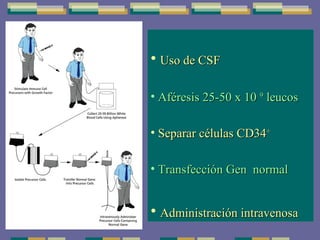

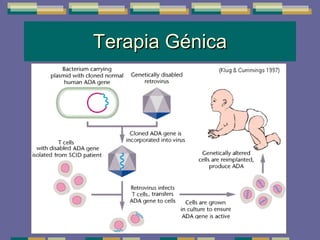


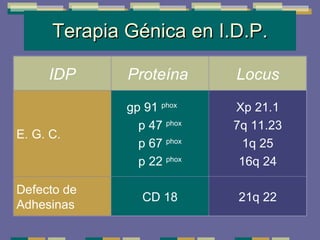
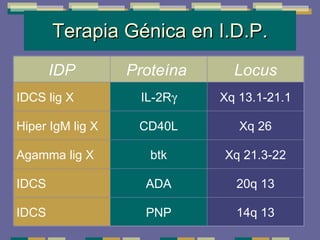
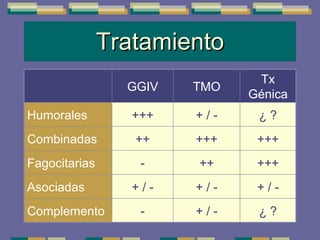


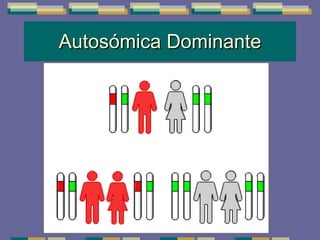

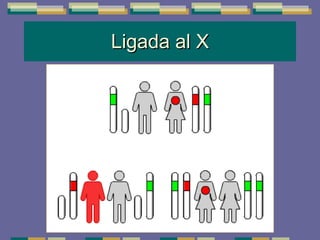

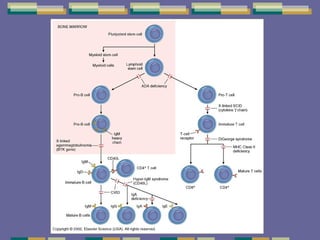



El documento trata sobre las inmunodeficiencias primarias. Explica que pueden ser de predominio humoral, celular, fagocítico o asociado al complemento. Describe varios tipos como la agammaglobulinemia, deficiencia de IgA, enfermedad de Bruton e inmunodeficiencia combinada severa. También cubre datos de alarma, estudios iniciales y avanzados para cada tipo, y formas de tratamiento como gammaglobulina e transplante de médula ósea.























































































