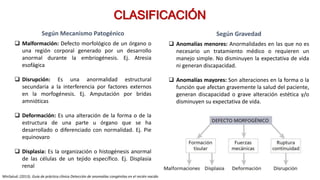
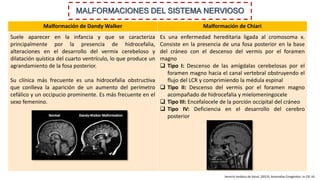
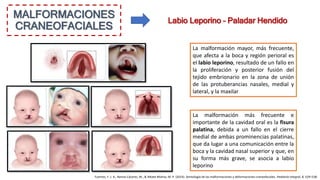
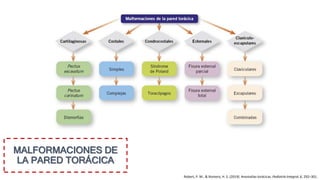
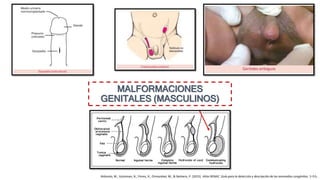
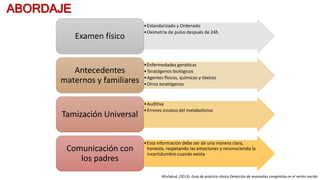
Este documento describe las malformaciones congénitas. Explica que son patologías caracterizadas por alteraciones estructurales o funcionales en un neonato debido a factores durante el desarrollo embrionario o fetal. Incluye varios tipos como malformaciones, disrupciones, deformidades, displasias y trastornos metabólicos. Las causas pueden ser genéticas, ambientales o desconocidas. Luego clasifica las anomalías según su mecanismo, gravedad y clínica. Describe varias malformaciones específicas como las