Descargado 753 veces

















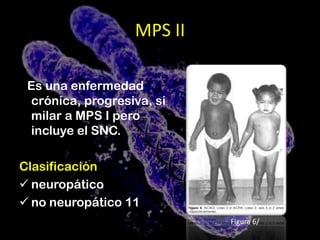



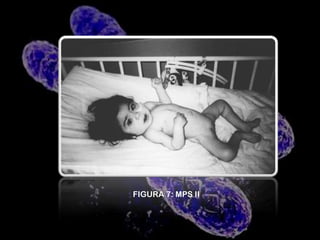











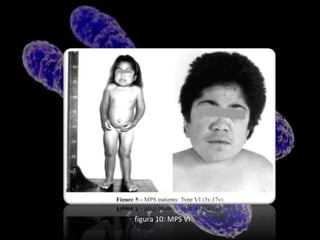





















Este documento describe la mucopolisacaridosis, un grupo de trastornos hereditarios causados por la falta de enzimas lisosomales que degradan los glicosaminoglicanos. Esto causa la acumulación progresiva de estos glicosaminoglicanos en los tejidos, lo que produce manifestaciones en múltiples sistemas. Se describen 7 tipos de mucopolisacaridosis dependiendo de la enzima deficiente y sus síntomas, así como opciones de tratamiento como el trasplante de células madre y terapia de re



































































