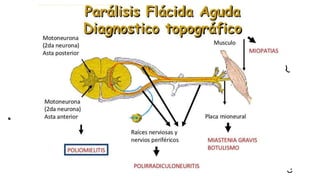
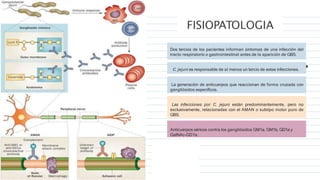
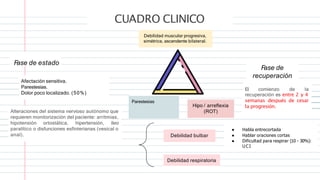
La parálisis flácida infantil se caracteriza por la pérdida súbita de fuerza y tono muscular en uno o más miembros en niños menores de 15 años. La causa principal es el síndrome de Guillain-Barré, una neuropatía autoinmune posinfecciosa que destruye la mielina de los nervios periféricos. Los síntomas incluyen debilidad muscular ascendente, ausencia de reflejos y, a veces, afectación de los nervios craneales y respiratorios. El diagnóstico se bas