Descargado 573 veces








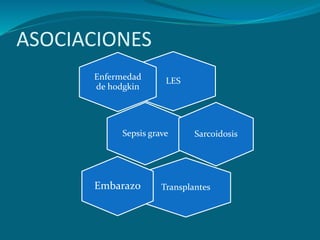

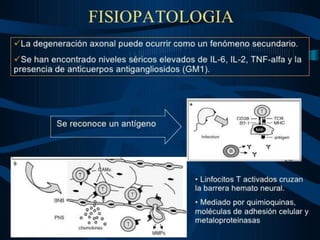
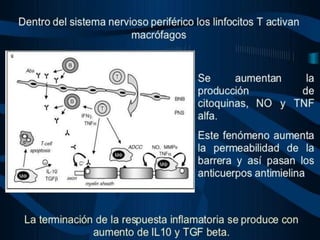
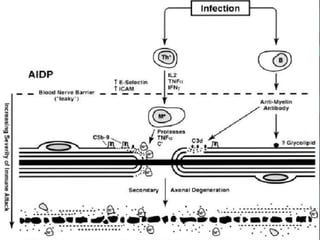
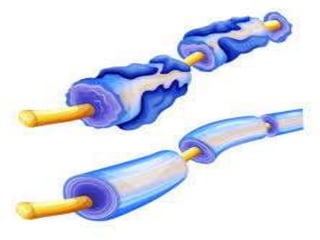























El documento define el síndrome de Guillain-Barré como una enfermedad autoinmune que causa debilidad muscular progresiva. Fue descubierto en 1916 por tres neurólogos que observaron una parálisis aguda arreflexica con recuperación espontánea. Se caracteriza por una proteinorraquia con celularidad normal en el líquido cefalorraquídeo. El tratamiento incluye inmunoglobulinas intravenosas o plasmaféresis para reducir la gravedad de los síntomas y acelerar la recuperación










































































